


- Home /
- Publicaciones de patentes /
- NUEVO TRATAMIENTO DEL CÁNCER COLORRECTAL
NUEVO TRATAMIENTO DEL CÁNCER COLORRECTAL
Patente nacional por "NUEVO TRATAMIENTO DEL CÁNCER COLORRECTAL"
Este registro ha sido solicitado por
FUNDACION UNIVERSITARIA SAN ANTONIO
a través del representanteMARIA DESAMPARADOS DIAZ PACHECO
Contacto
- Estado: Vigente
- País:
- España
- Fecha solicitud:
- 19/09/2021
- Número solicitud:
-
P202130871
- Número publicación:
-
ES2912350
- Fecha de concesión:
-
03/11/2023
- Inventores:
-
Persona física
- Datos del titular:
-
FUNDACION UNIVERSITARIA SAN ANTONIO
- Datos del representante:
-
MARIA DESAMPARADOS DIAZ PACHECO
- Clasificación Internacional de Patentes:
- A61K 31/185,A61P 35/00,A61P 35/04,A61P 7/02
- Clasificación Internacional de Patentes de la publicación:
- A61K 31/185,A61P 35/00,A61P 35/04,A61P 7/02
- Fecha de vencimiento:
Quiero registrar una patente

Reivindicaciones:
+ ES-2912350_B91.- Un compuesto de fórmula (I) o una sal, estereoisómero, tautómero, hidrato o solvato farmacéuticamente aceptable del mismo, para su uso en el tratamiento del cáncer colorrectal caracterizado por que el individuo presenta los niveles de hepsina elevados con respecto a los niveles normales y el cáncer colorrectal es no metastásico. 2.- Una composición que comprende el compuesto de fórmula (I) según la reivindicación anterior, para su uso en el tratamiento del cáncer colorrectal caracterizado por que el individuo presenta los niveles de hepsina elevados con respecto a los niveles normales y el cáncer colorrectal es no metastásico. 3.- La composición para su uso según la reivindicación anterior, que es una composición farmacéutica. 4.- La composición para su uso según cualquiera de las reivindicaciones 2-3, donde la composición además comprende excipientes y/o un vehículo farmacéuticamente aceptable. 5.- La composición para su uso según cualquiera de las reivindicaciones 3-4, que además comprende otro principio activo. 6.- Un kit de partes que comprende: a) Una parte A, que comprende un compuesto o una composición según cualquiera de las reivindicaciones 1-5, y b) Una parte B, que es un agente antineoplásico para su uso en el tratamiento del cáncer colorrectal donde el individuo presenta los niveles de hepsina elevados con respecto a los niveles normales y el cáncer colorrectal es no metastásico 7.- Un kit de partes que comprende: a) Una parte A, que comprende un compuesto o una composición según cualquiera de las reivindicaciones 1-5, y b) Una parte B, que es un agente antineoplásico para su uso en la prevención o el tratamiento de los eventos tromboembólicos venosos desarrollados como consecuencia de la terapia antineoplásica según la reivindicación 6.
+ ES-2912350_B21 Un compuesto de fórmula (I) o una sal, estereoisómero, tautómero, hidrato o solvato farmacéuticamente aceptable del mismo, para su uso en el tratamiento del cáncer colorrectal caracterizado por que el individuo presenta los niveles de hepsina elevados con respecto a los niveles normales y el cáncer colorrectal es no metastásico. 2.- Una composición que comprende el compuesto de fórmula (I) según la reivindicación anterior, para su uso en el tratamiento del cáncer colorrectal caracterizado por que el individuo presenta los niveles de hepsina elevados con respecto a los niveles normales y el cáncer colorrectal es no metastásico. 3.- La composición para su uso según la reivindicación anterior, que es una composición farmacéutica. 4.- La composición para su uso según cualquiera de las reivindicacione s 2-3, donde la composición además comprende excipientes y/o un vehículo farmacéuticamente aceptable. 5.- La composición para su uso según cualquiera de las reivindicaciones 3-4, que además comprende otro principio activo. 6.- Un kit de partes que comprende: a) Una parte A, que comprende un compuesto o una composición según cualquiera de las reivindicaciones 1-5, y b) Una parte B, que es un agente antineoplásico para su uso en el tratamiento del cáncer colorrectal donde el individuo presenta los niveles de hepsina elevados con respecto a los niveles normales y el cáncer colorrectal es no metastásico 7.- Un kit de partes que comprende: a) Una parte A, que comprende un compuesto o una composición según cualquiera de las reivindicaciones 1-5, y b) Una parte B, que es un agente antineoplásico para su uso en la prevención o el tratamiento de los eventos tromboembólicos venosos desarrollados como consecuencia de la terapia antineoplásica según la reivindicación 6.
+ ES-2912350_A11.- Un compuesto de fórmula (I) Fórmula (I) o una sal, estereoisómero, tautómero, hidrato o solvato farmacéuticamente aceptable del mismo, para la prevención y tratamiento del cáncer colorrectal. 2.- El compuesto para su uso según la reivindicación 1, donde el cáncer colorrectal es el cáncer colorrectal metastásico. 3.- El compuesto para su uso según cualquiera de las reivindicaciones 1-2, donde el individuo presenta los niveles de hepsina elevados con respecto a los niveles normales. 4.- Una composición que comprende un compuesto de fórmula (I) según la reivindicación anterior, para su uso según cualquiera de las reivindicaciones 1-3. 5.- La composición según la reivindicación anterior, que es una composición farmacéutica. 6.- La composición según cualquiera de las reivindicaciones 4-5, donde la composición además comprende excipientes y/o un vehículo farmacéuticamente aceptable. 7.- La composición según cualquiera de las reivindicaciones 4-6, que además comprende otro principio activo. 8.- Un kit de partes que comprende: a) Una parte A, que comprende un compuesto o una composición según cualquiera de las reivindicaciones 1-7, y b) Una parte B, que es un agente antineoplásico. 9.- El kit de partes según la reivindicación 8 para la prevención o el tratamiento de los eventos tromboembólicos venosos (ETEV) desarrollados como consecuencia de la terapia antineoplásica.
Los productos y servicios protegidos por este registro son:
A61K 31/185 - A61P 35/00 - A61P 35/04 - A61P 7/02
Descripciones:
+ ES-2912350_B9 Nuevo tratamiento del cáncer colorrectal CAMPO DE LA INVENCIÓN La presente invención pertenece al campo de la biomedicina, y más concretamente se refiere a un nuevo tratamiento para el cáncer colorrectal. ANTECEDENTES DE LA INVENCIÓN El cáncer colorrectal es el tercer cáncer más común diagnosticado tanto en hombres como en mujeres. Tiene lugar en el colon y recto y se desarrolla lentamente a partir de pólipos adenomatosos. El crecimiento excesivo de la mucosa del colon genera estos pólipos que tienen una morfología pedunculada o sésil. El desarrollo de cáncer colorrectal puede tener lugar por diferentes vías. Entre ellas se encuentra la aparición de lesiones serradas con apariencia del epitelio en dientes de sierra y que ocurre en el colon proximal. Esta vía se detecta en un tercio de todos los cánceres colorrectales. Con respecto a la secuencia adenoma-carcinoma, se ha propuesto que la progresión desde adenomas pequeños a grades adenomas o adenocarcinomas avanzados está mediada por múltiples rutas moleculares que incluyen la inestabilidad de microsatélites, la inestabilidad cromosómica y rutas epigenéticas (Jebelli, Asiyeh et al. Medicinal research reviews vol. 41, 1 (2021) : 395-434) . Es posible hacer una prevención precoz puesto que se ha estimado que al menos lleva 10 años que un pólipo se transforme en célula tumoral (0ines, Mari et al. "Epidemiology and risk factors of colorectal polyps." Best practice & research. Clinical gastroenterology vol. 31, 4 (2017) : 419-424) . El diagnóstico precoz mediante colonoscopia y detección de sangre oculta en heces, así como la mejora en los tratamientos ha contribuido a prolongar la supervivencia de los tumores colorrectales en la fase de curación, Sin embargo, la metástasis está presente en aproximadamente el 25% de los pacientes durante el diagnóstico y en total, el 50% de los pacientes con cáncer colorrectal desarrollará metástasis (Ohhara, Yoshihito et al. World journal of gastrointestinal oncology vol. 8, 9 (2016) : 642-55) . Debido a la heterogeneidad y naturaleza multifactorial del cáncer colorrectal han aumentado considerablemente en los últimos años estudios para identificar predictores clínicos y moleculares así como factores pronósticos con potencial para mejorar el manejo de los pacientes (Aran, Veronica et al. Clinical colorectal cancer vol. 15, 3 (2016) : 195-203) . Tanto la incidencia como la mortalidad del cáncer colorrectal ha disminuido tanto para hombres como para mujeres desde 1970 debido a la detección precoz. Sin embargo, aunque diferentes terapias estás disponibles para el cáncer colorrectal metastásico, los resultados no son óptimos debido a la heterogeneidad entre pacientes debido a características moleculares, respuesta a la terapia y presentación clínica (Linnekamp, Janneke F et al. Cancer research vol. 75, 2 (2015) : 245-9) . Los tratamientos más comunes de estos tumores se basan en la cirugía, la quimioterapia, terapias dirigidas y el tratamiento hormonal sustitutivo (Rejhová, A et al. European journal of medicinal chemistr y vol. 144 (2018) : 582-594) . Al menos el 50% de los pacientes con cáncer colorrectal recaen tras la resección quirúrgica y finalmente mueren de enfermedad metastásica. A pesar de la quimioterapia adyuvante postoperatoria en estos pacientes para disminuir el riesgo de recurrencia, el tratamiento adyuvante no da beneficios en supervivencia, especialmente en pacientes diagnosticados con estadío I (Aran, Veronica et al. Clinical colorectal cancer vol. 15, 3 (2016) : 195-203) . La terapia dirigida está basada en anticuerpos monoclonales y pequeños inhibidores moleculares, que selectivamente bloquean la proliferación celular mediante la intervención con ciertas moléculas y proteínas sobreexpresadas requeridas para la expansión del tumor (Jebelli, Asiyeh et al. Medicinal research reviews vol. 41, 1 (2021) : 395-434) . Las proteasas pericelulares han sido ampliamente implicadas en carcinogénesis, no solo porque actúan como enzimas capaces de degradar la matriz extracelular, permitiendo así a las células tumorales romper la membrana basal e invadir el tejido circundante, sino que son capaces de actuar como modificadores proteolíticos de factores de crecimiento y receptores activados por proteasas que son críticos para la activación de rutas de señalización tumoral (Tanabe, Lauren M, and Karin List. The FEBS journal vol. 284, 10 (2017) : 1421-1436) . Entre estas proteasas se encuentra hepsina, que pertenece a la super familia de serín proteasas transmembrana de tipo II (TTSP) . La Hepsina se encuentra sobreexpresada en cáncer de próstata y sus niveles elevados son indicativos de mal pronóstico y recaída tras prostatectomía radical (Sardana, Girish et al. Clinical chemistr y vol. 54, 12 (2008) : 1951-60.) . También se ha demostrado su sobreexpresión en cáncer de ovario (Miao, Jiangyong et al. International journal of cancer vol. 123, 9 (2008) : 2041-7) y cáncer de mama (Miao, Jiangyong et al. International journal of cancer vol. 123, 9 (2008) : 2041-7) . El desarrollo de eventos tromboembólicos venosos (ETEV) es un problema común en los tumores digestivos. La incidencia acumulada de ETEV en series modernas varía entre 12 20% (Posch, Florian et al. Thrombosis and haemostasis vol. 115, 4 (2016) : 817-26) , dependiendo de las disparidades en las características de los pacientes en cada estudio y el contexto en el que la evaluación es llevada a cabo. La trombogénesis se atribuye a una combinación de factores clínicos y biológicos, que incluyen la activación del sistema hemostático por parte del tumor y la toxicidad vascular de la quimioterapia. La trombosis se asocia con una mayor morbilidad y mortalidad en estos pacientes (Carmona-Bayonas, A et al. British journal of cancer vol. 116, 8 (2017) : 994-1001) y es uno de los eventos adversos más importantes en los ensayos clínicos de terapia anti-neoplásica. BREVE DESCRIPCIÓN DE LOS DIBUJOS Fig. 1. Fórmula de Suramina. Fig. 2. Pose resultante del docking molecular entre Suramina y hepsina. Fig. 3. Cálculo del valor de IC50 de Suramina sobre la actividad de Hepsina. Velocidad máxima de la actividad de Hepsina en presencia de dosis crecientes de Suramina. Fig. 4. Efecto de Suramina en la migración de células de cáncer colorrectal. Porcentaje de migración celular de las células Caco-2 y Caco-2-HPN en presencia o ausencia de Suramina. Fig. 5. Efecto de Suramina en la degración de gelatina de células de cáncer colorrectal. Porcentaje de degradación celular de las células Caco-2 y Caco-2-HPN en presencia o ausencia de Suramina. Fig. 6. Efecto de Suramina en la generación de trombina. Valores del potencial endógeno de trombina en células Caco-2 y Caco-2-HPN. Valores del tiempo hasta el inicio del pico (Lagtime) , yel tiempo en alcanzar el pico máximo (ttPeak) en las células Caco-2-HPN en presencia o ausencia de Suramina. Fig. 7. Efecto de Suramina en la proliferación de células de cáncer colorrectal. Porcentaje de células Caco-2 y Caco-2-HPN que están proliferando (EdU+) en presencia o ausencia de Suramina. Fig. 8. Efecto del pretratamiento con Suramina de células Caco-2-HPN (HPN-S) en la capacidad de invasión. DESCRIPCIÓN DE LA INVENCIÓN Los autores de la presente invención han demostrado que el Suramina reduce la migración de células de cáncer colorrectal, reduce la invasión de células de cáncer colorrectal, reduce la generación de trombina en plasma procedente de sujetos sanos tras la incubación con células de cáncer colorrectal y reduce la generación de trombina en plasma de pacientes con cáncer colorrectal con elevados niveles de hepsina en plasma. Han demostrado que el suramina, un fármaco utilizado para el tratamiento de la tripanosomiasis africana, la oncocercosis y en investigación para el tratamiento del cáncer de próstata (PMID: 15484217) , es inhibidor irreversible de hepsina. Por tanto, un primer aspecto de la invención se refiere al compuesto de fórmula (I) Fórmula (I) o cualquiera de sus sales, preferiblemente cualquier sal farmacéuticamente aceptable, ésteres, tautómeros, polimorfos, hidratos farmacéuticamente aceptables, o un isómero, profármacos, derivados, solvatos o análogos, o cualquiera de sus combinaciones, para su uso en el tratamiento del cáncer colorrectal caracterizado por que el individuo presenta los niveles de hepsina elevados con respecto a los niveles normales, y el cáncer colorrectal es cáncer no metastásico Como se puede observar en la Fig. 4, el Suramina es capaz de disminuir la migración de las células del cáncer colorrectal. Además, reduce de forma significativa la degradación de la gelatina de células Caco-2 tanto con expresión basal como con sobreexpresión de hepsina (Fig. 5) . Tal y como se muestra en la Fig. 6, el compuesto de la invención, reduce la generación de trombina en plasma de pacientes con cáncer colorrectal con elevados niveles de hepsina en plasma. En una realización particular, el compuesto de la invención se emplea para la prevención o tratamiento de los eventos tromboembólicos venosos (ETEV) , y preferiblemente de aquellos desarrollados como consecuencia de la terapia antineoplásica. Los compuestos de fórmula (I) , o sus sales o solvatos para uso de acuerdo con la invención están preferiblemente en forma farmacéuticamente aceptable o sustancialmente pura. Por forma farmacéuticamente aceptable se entiende, entre otras cosas, que tiene un nivel de pureza farmacéuticamente aceptable excluyendo los aditivos farmacéuticos normales tales como diluyentes y vehículos, y que no incluye ningún material considerado tóxico a niveles de dosificación normales. Los niveles de pureza de la sustancia farmacéutica están preferiblemente por encima del 50%, más preferiblemente por encima del 70%, lo más preferiblemente por encima del 90%. En una realización preferida, está por encima del 95% del compuesto de fórmula (I) o de su sal, estereoisómero o solvato farmacéuticamente aceptable. La invención también se refiere a usos según la invención de metabolitos de los compuestos descritos en la presente descripción. Un "metabolito" de un compuesto descrito en el presente documento es un derivado de ese compuesto que se forma cuando el compuesto se metaboliza. El término "metabolito activo" se refiere a un derivado biológicamente activo de un compuesto que se forma cuando el compuesto se metaboliza. El término "metabolizado", como se usa en este documento, se refiere a la suma de los procesos (que incluyen, pero no se limitan a, reacciones de hidrólisis y reacciones catalizadas por enzimas) mediante los cuales un organismo cambia una sustancia particular. Por tanto, las enzimas pueden producir alteraciones estructurales específicas en un compuesto. Después de entrar en el cuerpo, la mayoría de los medicamentos son sustratos para reacciones químicas que pueden cambiar us propiedades físicas y efectos biológicos. Estas conversiones metabólicas, que suelen afectar a la polaridad de los compuestos de la invención, alteran la forma en que los fármacos se distribuyen y excretan en el organismo. Sin embargo, en algunos casos, se requiere el metabolismo de un fármaco para obtener un efecto terapéutico. La invención también se refiere a profármacos de los compuestos para uso según la invención. El término "prodroga" o "profármaco" tal como aquí se utiliza incluye cualquier derivado de un compuesto de fórmula (I) , -por ejemplo y no limitativamente: ésteres (incluyendo ésteres de ácidos carboxílicos, ésteres de aminoácidos, ésteres de fosfato, ésteres de sulfonato de sales metálicas, etc.) , carbamatos, amidas, etc.- que al ser administrado a un individuo puede ser transformado directa o indirectamente en dicho compuesto de fórmula (I) en el mencionado individuo. Ventajosamente, dicho derivado es un compuesto que aumenta la biodisponibilidad del compuesto de fórmula (I) cuando se administra a un individuo o que potencia la liberación del compuesto de fórmula (I) en un compartimento biológico. La naturaleza de dicho derivado no es crítica siempre y cuando pueda ser administrado a un individuo y proporcione el compuesto de fórmula (I) , en un compartimento biológico de un individuo. La preparación de dicho profármaco puede llevarse a cabo mediante métodos convencionales conocidos por los expertos en la materia, que son capaces de liberar el compuesto de fórmula (I) como ingrediente activo cuando el profármaco se administra a un sujeto mamífero. La liberación del ingrediente activo ocurre in vivo. Los profármacos se pueden preparar mediante técnicas conocidas por los expertos en la técnica. Estas técnicas generalmente modifican los grupos funcionales apropiados en un compuesto dado. Sin embargo, estos grupos funcionales modificados regeneran grupos funcionales originales mediante manipulación rutinaria o in vivo. Los profármacos de los compuestos de la invención incluyen compuestos en los que se modifica un grupo hidroxi, amino, carboxílico o similar. Los ejemplos de profármacos incluyen, pero no se limitan a, ésteres. Los compuestos de fórmula general (I) y sus profármacos que contienen uno o varios centros quirales pueden estar presentes como racematos, mezclas diastereoméricas o isómeros individuales ópticamente activos. Los compuestos de la presente invención representados por la fórmula (I) pueden incluir isómeros, dependiendo de la presencia de enlaces múltiples, incluyendo isómeros ópticos o enantiómeros, dependiendo de la presencia de centros quirales. Los isómeros, enantiómeros o diastereoisómeros individuales y las mezclas de los mismos caen dentro del alcance de la presente invención, es decir, el término isómero también se refiere a cualquier mezcla de isómeros, como diastereómeros, racémicos, etc., incluso a sus isómeros ópticamente activos o las mezclas en distintas proporciones de los mismos. Los nantiómeros o diastereoisómeros individuales, así como sus mezclas, pueden separarse mediante técnicas convencionales. Composiciones y composiciones farmacéuticas El compuesto de fórmula (I) de la invención puede estar formando parte de una composición, preferiblemente de una composición farmacéutica. Otro aspecto de la invención se refiere a una composición que comprende el compuesto de fórmula (I) de la invención, de ahora en adelante composición de la invención, o una sal, estereoisómero, tautómero, hidrato o solvato farmacéuticamente aceptable del mismo, para la prevención y tratamiento del cáncer colorrectal caracterizado por que el individuo presenta los niveles de hepsina elevados con respecto a los niveles normales, y el cáncer colorrectal es cáncer no metastásico. En otra realización particular, se emplea el compuesto de la invención para la prevención o tratamiento de los eventos tromboembólicos venosos (ETEV) , y preferiblemente de aquellos desarrollados como consecuencia de la terapia antineoplásica. En otra realización de este aspecto, la composición de la invención además comprende otro principio activo. Más preferiblemente es una composición farmacéutica. En otra realización preferida además comprende vehículos farmacéuticamente aceptables. En otra realización preferida de este aspecto, la primera composición de la invención solo comprende como principio activo el compuesto de fórmula (I) , aunque puede comprender, además, excipientes y vehículos farmacéuticamente aceptables. El término "medicamento", tal y como se usa en esta memoria, hace referencia a cualquier sustancia usada para prevención, diagnóstico, alivio, tratamiento o curación de enfermedades en el hombre y los animales. La administración de los compuestos, composiciones o formas farmacéuticas de la presente invención puede ser realizada mediante cualquier método adecuado, como la infusión intravenosa y las vías oral, tópica o parenteral. La administración oral es la preferida por la conveniencia de los pacientes. La cantidad administrada de un compuesto de la presente invención dependerá de la relativa eficacia del compuesto elegido, la severidad de la enfermedad a tratar y el peso del paciente. Sin embargo, los compuestos de esta invención serán administrados una o más veces al día, por ejemplo 1, 2, 3 O 4 veces diarias, con una dosis total entre 0.1 y 1000 mg/Kg/día. Es importante tener en cuenta que puede ser necesario introducir variaciones en la dosis, dependiendo de la edad y de la condición del paciente, así como modificaciones en la vía de administración. "Composición farmacéutica", como se usa en el presente documento, se refiere a composiciones y entidades moleculares que son fisiológicamente tolerables y no producen típicamente una reacción alérgica o una reacción desfavorable similar a como trastornos gástricos, mareos y similares, cuando se administra a un humano o animal. Preferiblemente, el término "farmacéuticamente aceptable" significa que está aprobado por una agencia reguladora de un gobierno estatal o federal o está incluido en la Farmacopea de los Estados Unidos u otra farmacopea generalmente reconocida para uso en animales, y más particularmente en humanos. El término "excipiente" se refiere a un vehículo, diluyente o adyuvante que se administra con el ingrediente activo. Dichos excipientes farmacéuticos pueden ser líquidos estériles, como agua y aceites, incluidos los de origen petrolífero, animal, vegetal o sintético, como aceite de cacahuete, aceite de soja, aceite mineral, aceite de sésamo y similares. Como vehículos se utilizan preferiblemente agua o soluciones acuosas salinas y soluciones acuosas de dextrosa y glicerol, particularmente para soluciones inyectables. Los vehículos farmacéuticos adecuados se describen en "Remington's Phamaceutical Sciences" de EW Martin, 21a edición, 2005; o "Manual de excipientes farmacéuticos", Rowe CR; Paul JS; Marian EQ, sexta edición. Como se emplea aquí, el término "principio activo", "substancia activa", "substancia farmacéuticamente activa", "ingrediente activo" ó "ingrediente farmacéuticamente activo" significa cualquier componente que potencialmente proporcione una actividad farmacológica u otro efecto diferente en el diagnóstico, cura, mitigación, tratamiento, o prevención de una enfermedad, o que afecta a la estructura o función del cuerpo del hombre u otros animales. El término incluye aquellos componentes que promueven un cambio químico en la elaboración del fármaco y están presentes en el mismo de una forma modificada prevista que proporciona la actividad específica o el efecto. Las cantidades apropiadas de un compuesto de fórmula (I) como se define anteriormente, o una sal, estereoisómero farmacéuticamente aceptable o solvato del mismo se pueden formular con excipientes y/o vehículos farmacéuticamente aceptables para obtener una composición farmacéutica para uso en los usos médicos de la invención. Los vehículos farmacéuticamente aceptables adecuados incluyen, por ejemplo, agua, soluciones salinas, alcohol, aceites vegetales, polietilenglicoles, gelatina, lactosa, amilosa, estearato de magnesio, talco, tensioactivos, ácido silícico, parafina viscosa, aceite perfumado, monoglicéridos y diglicéridos. de ácidos grasos, ésteres de ácidos grasos, petroetrales, hidroximetilcelulosa, polivinilpirrolidona y similares. Las composiciones farmacéuticas que contienen el compuesto de fórmula (I) , como se definió anteriormente, o una sal, estereoisómero o solvato farmacéuticamente aceptable del mismo para su uso de acuerdo con la invención pueden ocurrir en cualquier forma farmacéutica de administración considerada apropiada para la vía de administración seleccionada, por ejemplo, por administración sistémica (por ejemplo, inyección intravenosa, subcutánea, intramuscular) , oral, parenteral o tópica, para lo cual incluirá los excipientes farmacéuticamente aceptables necesarios para la formulación del método de administración deseado. Además, también es posible administrar la composición que comprende el compuesto de fórmula (I) como se definió anteriormente, o una sal farmacéuticamente aceptable, estereoisómero o solvato del mismo para uso según la invención por vía intranasal o sublingual que permite la administración sistémica mediante un modo de administración no agresivo. Además, la administración intraventricular puede ser adecuada. Una vía de administración preferida es la oral. Cuando sea necesario, el compuesto de fórmula (I) como se definió anteriormente, o una sal, estereoisómero o solvato farmacéuticamente aceptable del mismo para su uso de acuerdo con la invención está comprendido en una composición que también incluye un agente solubilizante y un anestésico local para aliviar cualquier dolor en el lugar de la inyección. Generalmente, los ingredientes se suministran por separado o mezclados en forma de dosificación unitaria, por ejemplo, como un polvo liofilizado seco o un concentrado libre de agua en un recipiente herméticamente cerrado como una ampolla o bolsita que indica la cantidad de agente activo. Cuando la composición deba administrarse mediante infusión, se puede dispensar con una botella de infusión que contenga agua o solución salina estériles de calidad farmacéutica. En casos distintos de la administración intravenosa, la composición puede contener cantidades menores de agentes humectantes o emulsionantes, o agentes tamponantes del pH. La composición puede ser una solución líquida, suspensión, emulsión, gel, polímero o formulación de liberación sostenida. La composición se puede formular con aglutinantes y vehículos tradicionales, como se conoce en la técnica. Las formulaciones pueden incluir vehículos estándar tales como grados farmacéuticos de manitol, lactosa, almidón, estearato de magnesio, sacárido de sodio, celulosa, carbonato de magnesio, etc., vehículos inertes que tienen una funcionalidad bien establecida en la fabricación de productos farmacéuticos. Se conocen varios sistemas de administración y se pueden usar para administrar un compuesto de la presente invención que incluye la encapsulación en liposomas, micropartículas, microcápsulas y similares. Otro aspecto de la invención se refiere a una forma farmacéutica, de ahora en adelante forma farmacéutica de la invención, que comprende un compuesto de la invención (un compuesto de fórmula (I) , o la composición de la invención. En esta memoria se entiende por "forma farmacéutica" la mezcla de uno o más principios activos con o sin aditivos que presentan características físicas para su adecuada dosificación, conservación, administración y biodisponibilidad. Las formas de dosificación sólidas para administración oral pueden incluir cápsulas convencionales, cápsulas de liberación sostenida, comprimidos convencionales, comprimidos, comprimidos masticables, comprimidos sublinguales, efervescentes comprimidos, píldoras, suspensiones, polvos, gránulos y geles de liberación sostenida. En estas formas de dosificación sólidas, los compuestos activos se pueden mezclar con al menos un excipiente inerte como sacarosa, lactosa o almidón. Tales formas de dosificación también pueden comprender, como en la práctica normal, sustancias adicionales distintas de diluyentes inertes, por ejemplo, agentes lubricantes tales como estearato de magnesio. En el caso de cápsulas, comprimidos, comprimidos efervescentes y píldoras, las formas de dosificación también pueden comprender agentes tamponantes. Los comprimidos y las píldoras se pueden preparar con recubrimientos entéricos. Las formas de dosificación líquidas para administración oral pueden incluir emulsiones, soluciones, suspensiones, jarabes y elixires farmacéuticamente aceptables que contienen diluyentes inertes usados comúnmente en la técnica, tales como agua. Esas composiciones ambién pueden comprender adyuvantes tales como agentes humectantes, agentes emulsionantes y de suspensión y agentes edulcorantes, agentes aromatizantes y perfumantes. Las preparaciones inyectables, por ejemplo, suspensiones acuosas u oleaginosas, inyectables estériles se pueden formular de acuerdo con la técnica conocida usando agentes dispersantes, agentes humectantes y/o agentes de suspensión adecuados. Entre los vehículos y disolventes aceptables que se pueden utilizar se encuentran el agua, la solución de Ringer y la solución isotónica de cloruro de sodio. Los aceites estériles también se utilizan convencionalmente como disolventes o medios de suspensión. Para la administración tópica, los compuestos de la invención pueden formularse como cremas, geles, lociones, líquidos, pomadas, rocíe soluciones, dispersiones, barras sólidas, emulsiones, microemulsiones y similares que se pueden formular de acuerdo con métodos convencionales que utilizan excipientes adecuados, tales como, por ejemplo, emulsionantes, tensioactivos, agentes espesantes, agentes colorantes y combinaciones de dos o más de los mismos. Adicionalmente, el compuesto de fórmula (I) como se definió anteriormente, o una sal farmacéuticamente aceptable, estereoisómero o solvato del mismo para uso según la invención se pueden administrar en forma de parches transdérmicos o iontoforesis dispositivos. En una realización, los compuestos para uso según la invención se administran como un parche transdérmico, por ejemplo, en forma de parche transdérmico de liberación sostenida. Se conocen en la técnica parches transdérmicos adecuados. Varios sistemas de suministro de fármacos son conocidos y se pueden utilizar para administrar los agentes o composiciones para uso de acuerdo con la invención, incluyendo, por ejemplo, encapsulación en liposomas, microburbujas, emulsiones, micropartículas, microcápsulas y similares. La dosis requerida se puede administrar como una sola unidad o en una forma de liberación sostenida. Las formas de liberación sostenible y los materiales y métodos apropiados para su preparación se describen, por ejemplo, en " Modified-Release Drug Deliver y Technology ", Rathbone, MJ Hadgraft, J. y Roberts, MS (eds.) , Marcel Dekker, Inc., Nueva York (2002) , " Handbook of Pharmaceutical Controlled Release Technology ", Wise, DL (ed.) , Marcel Dekker, Inc. Nueva York, (2000) . En una realización de la invención, la forma administrable por vía ral de un compuesto para uso según la invención está en una forma de liberación sostenida que comprende además al menos un recubrimiento o matriz. El recubrimiento o matriz de liberación sostenida incluye, sin limitación, polímeros naturales, ceras, grasas, semisintéticos o sintéticos insolubles en agua, modificados, alcoholes grasos, ácidos grasos, plastificantes naturales semisintéticos o sintéticos, o una combinación de dos o más de ellos. Los recubrimientos entéricos se pueden aplicar usando procesos convencionales conocidos por los expertos en la técnica, como se describe en, por ejemplo, Johnson, JL, " Pharmaceutical tablet coating", Coatings Technology Handbook (Segunda edición) , Satas, D. y Tracton, AA (eds. ) , Marcel Dekker, Inc. Nueva York, (2001) , Carstensen, T., " Coating Tablets in Advanced Pharmaceutical Solids", Swarbrick, J. (ed.) , Marcel Dekker, Inc. Nueva York (2001) , 455-468. El término "prevención", "prevenir" o "prevenir", como se usa en el presente documento, se refiere a la administración de un compuesto de acuerdo con la invención o de un medicamento que comprende dicho compuesto a un sujeto que no ha sido diagnosticado como posiblemente tener una enfermedad, pero que normalmente se esperaría que la desarrolle o que tenga un mayor riesgo de padecerla. La prevención pretende evitar la aparición de dicha enfermedad. La prevención puede ser completa (por ejemplo, la ausencia total de una enfermedad) . La prevención también puede ser parcial, de modo que, por ejemplo, la aparición de una enfermedad en un sujeto es menor que la que habría ocurrido sin la administración del compuesto de la presente invención. La prevención también se refiere a la reducción de la susceptibilidad a una condición clínica. El término "tratamiento", como se usa en este documento, se refiere a cualquier tipo de terapia, que tiene como objetivo terminar, prevenir, mejorar o reducir la susceptibilidad a una condición clínica como se describe en este documento. En una realización preferida, el término tratamiento se refiere a un tratamiento profiláctico (es decir, una terapia para reducir la susceptibilidad a una afección clínica) , de un trastorno o afección como se define en el presente documento. Por tanto, "tratamiento", "tratar" y sus términos equivalentes se refieren a la obtención de un efecto farmacológico o fisiológico deseado, que cubre cualquier tratamiento de una afección o trastorno patológico en un mamífero, incluido un ser humano. El efecto puede ser profiláctico en términos de prevención total o parcial de un trastorno o síntoma del mismo y / o puede ser terapéutico en términos de una cura parcial o completa de un trastorno y / o efecto adverso atribuible al trastorno. Es decir, "tratamiento" incluye (1) evitar que el trastorno ocurra o reaparezca en un sujeto, (2) inhibir el trastorno, como detener su desarrollo, (3) detener o terminar el trastorno o, al menos, los síntomas asociados con el mismo, de modo que el huésped ya no sufra el trastorno o sus síntomas, como provocar la regresión del trastorno o sus síntomas, por ejemplo, al restaurar o reparar una función perdida, altante o defectuosa, o estimular un proceso ineficiente, o (4) aliviar, aliviar o mejorar el trastorno o los síntomas asociados con el mismo. El término "sujeto" como se usa en el presente documento, se refiere a cualquier sujeto, particularmente un sujeto mamífero, para el que se desea la terapia. Los sujetos mamíferos incluyen humanos, animales domésticos, animales de granja y zoológicos, deportes o animales de compañía como perros, gatos, cobayas, conejos, ratas, ratones, caballos, ganado, vacas, etc. En una realización preferida de la invención, el sujeto es un mamífero. En una realización más preferida de la invención, el sujeto es un ser humano. La administración de los compuestos, composiciones o formas farmacéuticas de la presente invención puede ser realizada mediante cualquier método adecuado, como la infusión intravenosa y las vías oral, tópica o parenteral. La administración oral es la preferida por la conveniencia de los pacientes. La cantidad administrada de un compuesto de la presente invención dependerá de la relativa eficacia del compuesto elegido, la severidad de la enfermedad a tratar y el peso del paciente. Sin embargo, los compuestos de esta invención serán administrados una o más veces al día, por ejemplo 1, 2, 3 ó 4 veces diarias, con una dosis total entre 0.1 y 1000 mg/Kg/día. Es importante tener en cuenta que puede ser necesario introducir variaciones en la dosis, dependiendo de la edad y de la condición del paciente, así como modificaciones en la vía de administración. A lo largo de la descripción y las reivindicaciones la palabra "comprende" y sus variantes no pretenden excluir otras características técnicas, aditivos, componentes o pasos. Para los expertos en la materia, otros objetos, ventajas y características de la invención se desprenderán en parte de la descripción y en parte de la práctica de la invención. Los siguientes ejemplos y figuras se proporcionan a modo de ilustración, y no se pretende que sean limitativos de la presente invención. EJEMPLOS DE LA INVENCIÓN Cribado virtual Con el objetivo de encontrar nuevos inhibidores para la hepsina se realizó un cribado virtual mediante la técnica de docking molecular a partir de la estructura cristalográfica de la proteína epositada en la Protein Data Bank con código 1P57 frente a la librería de compuestos DrugBank (https://go.drugbank.com/) en las coordenadas de su sitio catalítico, caracterizada por residuos HIS57, ASP102 y SER195. El docking molecular se realizó en un clúster de cómputo de alto rendimiento mediante el software Autodock Vina 1.1.2 ( http://vina.scripps.edu/) y para ello se convirtieron tanto la estructura de la proteína como la librería de ligandos DrugBank al formato pdbqt mediante MGLTools. Una vez terminados los cálculos se priorizaron los compuestos en función de docking score y se examinaron visualmente los 8 primeros, seleccionando el compuesto Suramina (código DrugBank DB04786 y primero en la lista) con un docking score de -12 Kcal/mol e interaciones hidrofóbicas con los residuos ALA-39, LEU-41, HIS-57, PRO-60, PHE-97, GLN-151, interacciónes mediante puente de hidrogeno con los residuos GLY-38, HIS-40, HIS-57, ASN-63, GLN-73, ASN-99, GLN-151, GLY-153, SER-195, SER-214, GLY-216, GLY-219, interacciones por "puentes de sal" con ARG-34, HIS-57 y una interacción "piStacking" con el residuo HIS-57 como se aprecia en la Figura 1. En dicha figura podemos observar que Suramina impide el acceso a la triada catalítica, representada en formato de superficie para sus tres residuos Suramina actúa como inhibidor irreversible de hepsina. Mediante un ensayo fluorogénico se determinó la capacidad de Suramina de inhibir la actividad proteolítica de hepsina. Hepsina es una serín proteasas capaz de hidrolizar al sustrato BOC-Gln-Arg-Arg-AMC (Bachem, Barcelona, Spain) , de manera que es posible registrar la emisión de fluorescencia mediante un lector de placas con longitudes de onda de excitación y emisión de 380 nm y 460 nm, respectivamente. Para ello, se incubó hepsina (0.05 j M) (R&D Systems, Madrid, Spain) en tampón 50 mM Tris-HCl, pH 9 buffer, con 200 |jM BOC-Gln-Arg-Arg-AMC y se registró la fluorescencia emitida durante 5 minutos. Para comprobar el efecto de Suramina, se llevó a cabo la misma reacción, pero incubando previamente hepsina con Suramina durante 1 hora a 37°C y en distintas concentraciones que oscilaron entre 0.13 y 10 j M. De esta forma se calculó el valor de IC50, o concentración de inhibidor a la que se alcanza la mitad de la velocidad máxima de hepsina en la hidrólisis de su sustrato, que fue de 0.66 j M. Suramina reduce la migración de células de cáncer colorrectal Una vez que las células alcanzan confluencia, forman una monocapa. Mediante la eliminación de una tira de células 300-500 ^m de anchura con una punta de pipetea de 200 ^l, es posible evaluar la capacidad de migración de las células y el efecto de Suramina en este proceso. La capacidad de migración se evalúa en función del porcentaje de área ocupada tras 48 horas de incubación y se cuantifica utilizando el software ImageJ. Los resultados muestran que Suramina a una concentración de 0, 66 ^M reduce de forma significativa la migración de células Caco-2, tanto con expresión basal como con sobreexpresión de hepsina, mediante transfección estable de un plásmido que contiene el gen HPN. Suramina reduce la invasión de células de cáncer colorrectal Para evaluar la invasión se realizó un ensayo en el que se evaluó la capacidad de las células para degradar una matriz de gelatina. Para ello, se mezcló gelatina al 0.2% y rodamina (Invitrogen, Life Technologies, Madrid, Spain) , en una ratio 1:55 en tampón NaCl 61 mM, borohidrato sódico 50 mM y después se dializó toda la noche en PBS. Se prepararon cubres con la mezcla preparada cubriéndolos y se fijó la matriz con glutaraldehido 0.5% durante 15 min. Los cubres se lavaron con PBS y se añadieron las suspensiones de células Caco-2 durante 72 horas. Posteriormente se fijaron las células con formaldehído 3.7% y se incubó con phalloidin faloidin?? (0, 01 mg/ml) y ProLongGold Antifade con DAPI (4', 6-diamino-2-phenylindole; ThermoFisher) . Las imágenes se tomaron con un microscopio confocal SP8 LEICA y se analizaron con ImageJ. Los resultados mostraron que Suramina, a una concentración de 0, 66 ^M, reduce de forma significativa la degradación de la gelatina de células Caco-2 tanto con expresión basal como con sobreexpresión de hepsina. Suramina reduce la generación de trombina Puesto que hepsina activa al FVII y, por tanto, la cascada de la coagulación, podría contribuir al estado de hipercoagulabilidad de los pacientes con cáncer colorrectal con niveles elevados de hepsina. Para comprobar la hipótesis, se incubó plasma procedente de 20 sujetos sanos, a los que se extrajo sangre citratada 3.8% mediante venopunción, con las células Caco-2 con sobreexpresión de hepsina. El plasma se centrifugó previamente a 2500 g 20 minutos. Para el ensayo de generación de trombina, se retiró el plasma incubado y se incubó con el reactivo LOW® (factor tisular: 1 pmol; fosfolípidos: 4 ^mol; Diagnostica Stago) en una placa de 96 pocillo. Todas las muestras se prepararon en duplicado. La coagulación en estas muestras se inició añadiendo cloruro cálcico en un tampón que contenía el sustrato fluorogénico FluCa-kit reagent® (Diagnostica Stago) . Para cada muestra individual se utilizó un calibrador de rombina. La fluorescencia se registró durante 60 min en un fluorímetro Fluoroskan Ascent (Thermolab Systems) y los datos se analizaron utilizando el software Thrombinoscope™ (version 5.0.0.742; Diagnostica Stago) . Se analizaron los siguientes parámetros: (a) lag-time, que indica el inicio de la fase de generación de trombina; (b) tiempo hasta alcanzar la máxima concentración de trombina (ttPeak) ; (c) concentración máxima de trombina (Peak) ; (d) índice de frecuencia media (MRI) de la fase de propagación de la generación de trombina calculada por la fórmula Peak/ (ttPeak - lag-time) y se expresa en nM/min; y (e) potencial endógeno de trombina (ETP) que muestra la actividad enzimática de trombina evaluada como el área bajo la curva. Los resultados muestran que Suramina reduce el potencial endógeno de trombina (ETP) , el pico máximo (peak) y la velocidad en alcanzar el pico mientras que provoca un incremento en el tiempo de inicio de la fase de generación de trombina (lag-time) y el tiempo en alcanzar el pico. Tabla 1. Valores promedios del test de generación de trombina. Suramina no afecta ni a la muerte celular, ni a la proliferación celular Se quiso evaluar si la menor migración o invasión celular podría deberse a una mayor muerte, o a una menor proliferación celular. Para ello se incubaron las células (5x105 cells/ml) en placas de 6 pocillos en presencia o ausencia de Suramina tras 48 h. El tipo de muerte celular se determinó mediante el ensayo de anexina V/7-ADD (Anexin V Apoptosis Detection Kit FITC; eBiosciences, Thermo Fisher, Karlsruhe, Alemania; 7-ADD BD-Biosciences) siguiendo las instrucciones del fabricante. Para evaluar la actividad proliferativa de las células Caco-2, se añadió 5-etinil-2'-desoxiuridina (EdU) (Thermo Fisher Scientific) durante 48 h. Luego, se usaron alícuotas de las células tumorales para determinar el porcentaje de células positivas, es decir, Caco-2 o Caco-2-HPN-EdU+, detectadas por reacción de acoplamiento de azida luorescente con EdU de acuerdo con el protocolo del fabricante (Click-iT; Thermo Fisher Scientific) . Los resultados mostraron que Suramina no afecta ni a la muerte (porcentajes entorno al 5-10 %) ni a la proliferación celular. Suramina inhibe la invasión de las células de cáncer colorrectal en modelos de xenotrasplante de larvas de pez cebra Se obtuvieron peces cebra wild type (Danio rerio H. Cypriniformes, Cyprinidae) del Zebrafish International Resource Center (ZIRC, Oregon, EE. UU.) Los peces roya9/a9; nacrew2/w2 (casper) transparentes se obtuvieron como ya se ha descrito previamente en la bibliografía [R.M. White, A. Sessa, C. Burke, T. Bowman, J. LeBlanc, C. Ceol, et al. Transparent adult zebrafish as a tool for in vivo transplantation analysis. Cell Stem Cell 2008;2:183-9]. Los peces se aparearon, clasificaron, criaron y procesaron como se describe en el manual del pez cebra [M. Westerfield. The zebrafish book: a guide for the laborator y use of zebrafish. Http//Zfin Org/Zf_info/Zfbook/Zfbk Html 2000.]. Los huevos de pez cebra fertilizados se obtuvieron del desove natural y se mantuvieron en nuestras instalaciones siguiendo las prácticas estándar de cría. Los animales se mantuvieron en un ciclo de luz / oscuridad de 12 h a 28 ° C. Las larvas de pez cebra se anestesiaron con 0.16 mg/ml ethyl 3-aminobenzoato (tricaine; Sigma-Aldrich) . Las células Caco-2 y Caco-2-HPN se cultivaron en presencia y ausencia de 0, 66 ^M de suramina durante 48 h, luego se disgregaron y marcaron con 1, 1'-di-octa-decil-3, 3, 3 ', 3'-perclorato de tetra-metil-indo-carbo-cianina (Dil, ThermoFisher) y finalmente se resuspendieron en un tampón que contenía suero bovino fetal al 5% en PBS. La inyección de las células en embriones se realizó mediante la inyección de 200 células/embrión en el saco vitelino de larvas de pez cebra wild type o Casper 48 horas después de la fertilización y después de 5 días a 35°C, las larvas se analizaron mediante microscopía de fluorescencia para comprobar la diseminación de las células tumorales. La puntuación de la invasión de células Caco-2 y Caco-2-HPN se calculó como el porcentaje de larvas con invasión de células tumorales con respecto al total de larvas analizadas. El pretratamiento de las células Caco-2-HPN (CACO HPN) con suramina (HPN + S) redujo su capacidad de invasión a los niveles encontrados en las células parentales (CACO WT) (Figura 8) , lo que confirma los estudios in vitro y muestra además su potencial terapéutico para el tratamiento del cáncer colorrectal.
+ ES-2912350_B2 Nuevo tratamiento del cáncer colorrectal CAMPO DE LA INVENCIÓN La presente invención pertenece al campo de la biomedicina, y más concretamente se refiere a un nuevo tratamiento para el cáncer colorrectal. ANTECEDENTES DE LA INVENCIÓN El cáncer colorrectal es el tercer cáncer más común diagnosticado tanto en hombres como en mujeres. Tiene lugar en el colon y recto y se desarrolla lentamente a partir de pólipos adenomatosos. El crecimiento excesivo de la mucosa del colon genera estos pólipos que tienen una morfología pedunculada o sésil. El desarrollo de cáncer colorrectal puede tener lugar por diferentes vías. Entre ellas se encuentra la aparición de lesiones serradas con apariencia del epitelio en dientes de sierra y que ocurre en el colon proximal. Esta vía se detecta en un tercio de todos los cánceres colorrectales. Con respecto a la secuencia adenoma-carcinoma, se ha propuesto que la progresión desde adenomas pequeños a grades adenomas o adenocarcinomas avanzados está mediada por múltiples rutas moleculares que incluyen la inestabilidad de microsatélites, la inestabilidad cromosómica y rutas epigenéticas (Jebelli, Asiyeh et al. Medicinal research reviews vol. 41, 1 (2021) : 395-434) . Es posible hacer una prevención precoz puesto que se ha estimado que al menos lleva 10 años que un pólipo se transforme en célula tumoral (0ines, Mari et al. "Epidemiology and risk factors of colorectal polyps." Best practice & research. Clinical gastroenterology vol. 31, 4 (2017) : 419-424) . El diagnóstico precoz mediante colonoscopia y detección de sangre oculta en heces, así como la mejora en los tratamientos ha contribuido a prolongar la supervivencia de los tumores colorrectales en la fase de curación, Sin embargo, la metástasis está presente en aproximadamente el 25% de los pacientes durante el diagnóstico y en total, el 50% de los pacientes con cáncer colorrectal desarrollará metástasis (Ohhara, Yoshihito et al. World journal of gastrointestinal oncology vol. 8, 9 (2016) : 642-55) . Debido a la heterogeneidad y naturaleza multifactorial del cáncer colorrectal han aumentado considerablemente en los últimos años estudios para identificar predictores clínicos y moleculares así como factores pronósticos con potencial para mejorar el manejo de los pacientes (Aran, Veronica et al. Clinical colorectal cancer vol. 15, 3 (2016) : 195-203) . Tanto la incidencia como la mortalidad del cáncer colorrectal ha disminuido tanto para hombres como para mujeres desde 1970 debido a la detección precoz. Sin embargo, aunque diferentes terapias estás disponibles para el cáncer colorrectal metastásico, los resultados no son óptimos debido a la heterogeneidad entre pacientes debido a características moleculares, respuesta a la terapia y presentación clínica (Linnekamp, Janneke F et al. Cancer research vol. 75, 2 (2015) : 245-9) . Los tratamientos más comunes de estos tumores se basan en la cirugía, la quimioterapia, terapias dirigidas y el tratamiento hormonal sustitutivo (Rejhová, A et al. European journal of medicinal chemistr y vol. 144 (2018) : 582-594) . Al menos el 50% de los pacientes con cáncer colorrectal recaen tras la resección quirúrgica y finalmente mueren de enfermedad metastásica. A pesar de la quimioterapia adyuvante postoperatoria en estos pacientes para disminuir el riesgo de recurrencia, el tratamiento adyuvante no da beneficios en supervivencia, especialmente en pacientes diagnosticados con estadío I (Aran, Veronica et al. Clinical colorectal cancer vol. 15, 3 (2016) : 195-203) . La terapia dirigida está basada en anticuerpos monoclonales y pequeños inhibidores moleculares, que selectivamente bloquean la proliferación celular mediante la intervención con ciertas moléculas y proteínas sobreexpresadas requeridas para la expansión del tumor (Jebelli, Asiyeh et al. Medicinal research reviews vol. 41, 1 (2021) : 395-434) . Las proteasas pericelulares han sido ampliamente implicadas en carcinogénesis, no solo porque actúan como enzimas capaces de degradar la matriz extracelular, permitiendo así a las células tumorales romper la membrana basal e invadir el tejido circundante, sino que son capaces de actuar como modificadores proteolíticos de factores de crecimiento y receptores activados por proteasas que son críticos para la activación de rutas de señalización tumoral (Tanabe, Lauren M, and Karin List. The FEBS journal vol. 284, 10 (2017) : 1421-1436) . Entre estas proteasas se encuentra hepsina, que pertenece a la super familia de serín proteasas transmembrana de tipo II (TTSP) . La Hepsina se encuentra sobreexpresada en cáncer de próstata y sus niveles elevados son indicativos de mal pronóstico y recaída tras prostatectomía radical (Sardana, Girish et al. Clinical chemistr y vol. 54, 12 (2008) : 1951-60.) . También se ha demostrado su sobreexpresión en cáncer de ovario (Miao, Jiangyong et al. International journal of cancer vol. 123, 9 (2008) : 2041-7) y cáncer de mama (Miao, Jiangyong et al. International journal of cancer vol. 123, 9 (2008) : 2041-7) . El desarrollo de eventos tromboembólicos venosos (ETEV) es un problema común en los tumores digestivos. La incidencia acumulada de ETEV en series modernas varía entre 12 20% (Posch, Florian et al. Thrombosis and haemostasis vol. 115, 4 (2016) : 817-26) , dependiendo de las disparidades en las características de los pacientes en cada estudio y el contexto en el que la evaluación es llevada a cabo. La trombogénesis se atribuye a una combinación de factores clínicos y biológicos, que incluyen la activación del sistema hemostático por parte del tumor y la toxicidad vascular de la quimioterapia. La trombosis se asocia con una mayor morbilidad y mortalidad en estos pacientes (Carmona-Bayonas, A et al. British journal of cancer vol. 116, 8 (2017) : 994-1001) y es uno de los eventos adversos más importantes en los ensayos clínicos de terapia anti-neoplásica. BREVE DESCRIPCIÓN DE LOS DIBUJOS Fig. 1. Fórmula de Suramina. Fig. 2. Pose resultante del docking molecular entre Suramina y hepsina. Fig. 3. Cálculo del valor de IC50 de Suramina sobre la actividad de Hepsina. Velocidad máxima de la actividad de Hepsina en presencia de dosis crecientes de Suramina. Fig. 4. Efecto de Suramina en la migración de células de cáncer colorrectal. Porcentaje de migración celular de las células Caco-2 y Caco-2-HPN en presencia o ausencia de Suramina. Fig. 5. Efecto de Suramina en la degración de gelatina de células de cáncer colorrectal. Porcentaje de degradación celular de las células Caco-2 y Caco-2-HPN en presencia o ausencia de Suramina. Fig. 6. Efecto de Suramina en la generación de trombina. Valores del potencial endógeno de trombina en células Caco-2 y Caco-2-HPN. Valores del tiempo hasta el inicio del pico (Lagtime) , yel tiempo en alcanzar el pico máximo (ttPeak) en las células Caco-2-HPN en presencia o ausencia de Suramina. Fig. 7. Efecto de Suramina en la proliferación de células de cáncer colorrectal. Porcentaje de células Caco-2 y Caco-2-HPN que están proliferando (EdU+) en presencia o ausencia de Suramina. Fig. 8. Efecto del pretratamiento con Suramina de células Caco-2-HPN (HPN-S) en la capacidad de invasión. DESCRIPCIÓN DE LA INVENCIÓN Los autores de la presente invención han demostrado que el Suramina reduce la migración de células de cáncer colorrectal, reduce la invasión de células de cáncer colorrectal, reduce la generación de trombina en plasma procedente de sujetos sanos tras la incubación con células de cáncer colorrectal y reduce la generación de trombina en plasma de pacientes con cáncer colorrectal con elevados niveles de hepsina en plasma. Han demostrado que el suramina, un fármaco utilizado para el tratamiento de la tripanosomiasis africana, la oncocercosis y en investigación para el tratamiento del cáncer de próstata (PMID: 15484217) , es inhibidor irreversible de hepsina. Por tanto, un primer aspecto de la invención se refiere al compuesto de fórmula (I) Fórmula (I) o cualquiera de sus sales, preferiblemente cualquier sal farmacéuticamente aceptable, ésteres, tautómeros, polimorfos, hidratos farmacéuticamente aceptables, o un isómero, profármacos, derivados, solvatos o análogos, o cualquiera de sus combinaciones, para su uso en el tratamiento del cáncer colorrectal caracterizado por que el individuo presenta los niveles de hepsina elevados con respecto a los niveles normales, y el cáncer colorrectal es cáncer no metastásico Como se puede observar en la Fig. 4, el Suramina es capaz de disminuir la migración de las células del cáncer colorrectal. Además, reduce de forma significativa la degradación de la gelatina de células Caco-2 tanto con expresión basal como con sobreexpresión de hepsina (Fig. 5) . Tal y como se muestra en la Fig. 6, el compuesto de la invención, reduce la generación de trombina en plasma de pacientes con cáncer colorrectal con elevados niveles de hepsina en plasma. En una realización particular, el compuesto de la invención se emplea para la prevención o tratamiento de los eventos tromboembólicos venosos (ETEV) , y preferiblemente de aquellos desarrollados como consecuencia de la terapia antineoplásica. Los compuestos de fórmula (I) , o sus sales o solvatos para uso de acuerdo con la invención están preferiblemente en forma farmacéuticamente aceptable o sustancialmente pura. Por forma farmacéuticamente aceptable se entiende, entre otras cosas, que tiene un nivel de pureza farmacéuticamente aceptable excluyendo los aditivos farmacéuticos normales tales como diluyentes y vehículos, y que no incluye ningún material considerado tóxico a niveles de dosificación normales. Los niveles de pureza de la sustancia farmacéutica están preferiblemente por encima del 50%, más preferiblemente por encima del 70%, lo más preferiblemente por encima del 90%. En una realización preferida, está por encima del 95% del compuesto de fórmula (I) o de su sal, estereoisómero o solvato farmacéuticamente aceptable. La invención también se refiere a usos según la invención de metabolitos de los compuestos descritos en la presente descripción. Un "metabolito" de un compuesto descrito en el presente documento es un derivado de ese compuesto que se forma cuando el compuesto se metaboliza. El término "metabolito activo" se refiere a un derivado biológicamente activo de un compuesto que se forma cuando el compuesto se metaboliza. El término "metabolizado", como se usa en este documento, se refiere a la suma de los procesos (que incluyen, pero no se limitan a, reacciones de hidrólisis y reacciones catalizadas por enzimas) mediante los cuales un organismo cambia una sustancia particular. Por tanto, las enzimas pueden producir alteraciones estructurales específicas en un compuesto. Después de entrar en el cuerpo, la mayoría de los medicamentos son sustratos para reacciones químicas que pueden cambiar sus propiedades físicas y efectos biológicos. Estas conversiones metabólicas, que suelen fectar a la polaridad de los compuestos de la invención, alteran la forma en que los fármacos se distribuyen y excretan en el organismo. Sin embargo, en algunos casos, se requiere el metabolismo de un fármaco para obtener un efecto terapéutico. La invención también se refiere a profármacos de los compuestos para uso según la invención. El término "prodroga" o "profármaco" tal como aquí se utiliza incluye cualquier derivado de un compuesto de fórmula (I) , -por ejemplo y no limitativamente: ésteres (incluyendo ésteres de ácidos carboxílicos, ésteres de aminoácidos, ésteres de fosfato, ésteres de sulfonato de sales metálicas, etc.) , carbamatos, amidas, etc.- que al ser administrado a un individuo puede ser transformado directa o indirectamente en dicho compuesto de fórmula (I) en el mencionado individuo. Ventajosamente, dicho derivado es un compuesto que aumenta la biodisponibilidad del compuesto de fórmula (I) cuando se administra a un individuo o que potencia la liberación del compuesto de fórmula (I) en un compartimento biológico. La naturaleza de dicho derivado no es crítica siempre y cuando pueda ser administrado a un individuo y proporcione el compuesto de fórmula (I) , en un compartimento biológico de un individuo. La preparación de dicho profármaco puede llevarse a cabo mediante métodos convencionales conocidos por los expertos en la materia, que son capaces de liberar el compuesto de fórmula (I) como ingrediente activo cuando el profármaco se administra a un sujeto mamífero. La liberación del ingrediente activo ocurre in vivo. Los profármacos se pueden preparar mediante técnicas conocidas por los expertos en la técnica. Estas técnicas generalmente modifican los grupos funcionales apropiados en un compuesto dado. Sin embargo, estos grupos funcionales modificados regeneran grupos funcionales originales mediante manipulación rutinaria o in vivo. Los profármacos de los compuestos de la invención incluyen compuestos en los que se modifica un grupo hidroxi, amino, carboxílico o similar. Los ejemplos de profármacos incluyen, pero no se limitan a, ésteres. Los compuestos de fórmula general (I) y sus profármacos que contienen uno o varios centros quirales pueden estar presentes como racematos, mezclas diastereoméricas o isómeros individuales ópticamente activos. Los compuestos de la presente invención representados por la fórmula (I) pueden incluir isómeros, dependiendo de la presencia de enlaces múltiples, incluyendo isómeros ópticos o enantiómeros, dependiendo de la presencia de centros quirales. Los isómeros, enantiómeros o diastereoisómeros individuales y las mezclas de los mismos caen dentro del alcance de la presente invención, es decir, el término isómero también se refiere a cualquier mezcla de isómeros, como diastereómeros, racémicos, etc., incluso a sus isómeros ópticamente activos o las mezclas en distintas proporciones de los mismos. Los nantiómeros o diastereoisómeros individuales, así como sus mezclas, pueden separarse mediante técnicas convencionales. Composiciones y composiciones farmacéuticas El compuesto de fórmula (I) de la invención puede estar formando parte de una composición, preferiblemente de una composición farmacéutica. Otro aspecto de la invención se refiere a una composición que comprende el compuesto de fórmula (I) de la invención, de ahora en adelante composición de la invención, o una sal, estereoisómero, tautómero, hidrato o solvato farmacéuticamente aceptable del mismo, para la prevención y tratamiento del cáncer colorrectal caracterizado por que el individuo presenta los niveles de hepsina elevados con respecto a los niveles normales, y el cáncer colorrectal es cáncer no metastásico. En otra realización particular, se emplea el compuesto de la invención para la prevención o tratamiento de los eventos tromboembólicos venosos (ETEV) , y preferiblemente de aquellos desarrollados como consecuencia de la terapia antineoplásica. En otra realización de este aspecto, la composición de la invención además comprende otro principio activo. Más preferiblemente es una composición farmacéutica. En otra realización preferida además comprende vehículos farmacéuticamente aceptables. En otra realización preferida de este aspecto, la primera composición de la invención solo comprende como principio activo el compuesto de fórmula (I) , aunque puede comprender, además, excipientes y vehículos farmacéuticamente aceptables. El término "medicamento", tal y como se usa en esta memoria, hace referencia a cualquier sustancia usada para prevención, diagnóstico, alivio, tratamiento o curación de enfermedades en el hombre y los animales. La administración de los compuestos, composiciones o formas farmacéuticas de la presente invención puede ser realizada mediante cualquier método adecuado, como la infusión intravenosa y las vías oral, tópica o parenteral. La administración oral es la preferida por la conveniencia de los pacientes. La cantidad administrada de un compuesto de la presente invención dependerá de la relativa eficacia del compuesto elegido, la severidad de la enfermedad a tratar y el peso del paciente. Sin embargo, los compuestos de esta invención serán administrados una o más veces al día, por ejemplo 1, 2, 3 O 4 veces diarias, con una dosis total entre 0.1 y 1000 mg/Kg/día. Es importante tener en cuenta que puede ser necesario introducir variaciones en la dosis, dependiendo de la edad y de la condición del paciente, así como modificaciones en la vía de administración. "Composición farmacéutica", como se usa en el presente documento, se refiere a composiciones y entidades moleculares que son fisiológicamente tolerables y no producen típicamente una reacción alérgica o una reacción desfavorable similar a como trastornos gástricos, mareos y similares, cuando se administra a un humano o animal. Preferiblemente, el término "farmacéuticamente aceptable" significa que está aprobado por una agencia reguladora de un gobierno estatal o federal o está incluido en la Farmacopea de los Estados Unidos u otra farmacopea generalmente reconocida para uso en animales, y más particularmente en humanos. El término "excipiente" se refiere a un vehículo, diluyente o adyuvante que se administra con el ingrediente activo. Dichos excipientes farmacéuticos pueden ser líquidos estériles, como agua y aceites, incluidos los de origen petrolífero, animal, vegetal o sintético, como aceite de cacahuete, aceite de soja, aceite mineral, aceite de sésamo y similares. Como vehículos se utilizan preferiblemente agua o soluciones acuosas salinas y soluciones acuosas de dextrosa y glicerol, particularmente para soluciones inyectables. Los vehículos farmacéuticos adecuados se describen en "Remington's Pharmaceutical Sciences" de EW Martin, 21a edición, 2005; o "Manual de excipientes farmacéuticos", Rowe CR; Paul JS; Marian EQ, sexta edición. Como se emplea aquí, el término "principio activo", "substancia activa", "substancia farmacéuticamente activa", "ingrediente activo" ó "ingrediente farmacéuticamente activo" significa cualquier componente que potencialmente proporcione una actividad farmacológica u otro efecto diferente en el diagnóstico, cura, mitigación, tratamiento, o prevención de una enfermedad, o que afecta a la estructura o función del cuerpo del hombre u otros animales. El término incluye aquellos componentes que promueven un cambio químico en la elaboración del fármaco y están presentes en el mismo de una forma modificada prevista que proporciona la actividad específica o el efecto. Las cantidades apropiadas de un compuesto de fórmula (I) como se define anteriormente, o una sal, estereoisómero farmacéuticamente aceptable o solvato del mismo se pueden formular con excipientes y/o vehículos farmacéuticamente aceptables para obtener una composición farmacéutica para uso en los usos médicos de la invención. Los vehículos farmacéuticamente aceptables adecuados incluyen, por ejemplo, agua, soluciones salinas, alcohol, aceites vegetales, polietilenglicoles, gelatina, lactosa, amilosa, estearato de magnesio, talco, tensioactivos, ácido silícico, parafina viscosa, aceite perfumado, monoglicéridos y diglicéridos. de ácidos grasos, ésteres de ácidos grasos, petroetrales, hidroximetilcelulosa, polivinilpirrolidona y similares. Las composiciones farmacéuticas que contienen el compuesto de fórmula (I) , como se definió anteriormente, o una sal, estereoisómero o solvato farmacéuticamente aceptable del mismo para su uso de acuerdo con la invención pueden ocurrir en cualquier forma farmacéutica de administración considerada apropiada para la vía de administración seleccionada, por ejemplo, por administración sistémica (por ejemplo, inyección intravenosa, subcutánea, intramuscular) , oral, parenteral o tópica, para lo cual incluirá los excipientes farmacéuticamente aceptables necesarios para la formulación del método de administración deseado. Además, también es posible administrar la composición que comprende el compuesto de fórmula (I) como se definió anteriormente, o una sal farmacéuticamente aceptable, estereoisómero o solvato del mismo para uso según la invención por vía intranasal o sublingual que permite la administración sistémica mediante un modo de administración no agresivo. Además, la administración intraventricular puede ser adecuada. Una vía de administración preferida es la oral. Cuando sea necesario, el compuesto de fórmula (I) como se definió anteriormente, o una sal, estereoisómero o solvato farmacéuticamente aceptable del mismo para su uso de acuerdo con la invención está comprendido en una composición que también incluye un agente solubilizante y un anestésico local para aliviar cualquier dolor en el lugar de la inyección. Generalmente, los ingredientes se suministran por separado o mezclados en forma de dosificación unitaria, por ejemplo, como un polvo liofilizado seco o un concentrado libre de agua en un recipiente herméticamente cerrado como una ampolla o bolsita que indica la cantidad de agente activo. Cuando la composición deba administrarse mediante infusión, se puede dispensar con una botella de infusión que contenga agua o solución salina estériles de calidad farmacéutica. En casos distintos de la administración intravenosa, la composición puede contener cantidades menores de agentes humectantes o emulsionantes, o agentes tamponantes del pH. La composición puede ser una solución líquida, suspensión, emulsión, gel, polímero o formulación de liberación sostenida. La composición se puede formular con aglutinantes y vehículos tradicionales, como se conoce en la técnica. Las formulaciones pueden incluir vehículos estándar tales como grados farmacéuticos de manitol, lactosa, almidón, estearato de magnesio, sacárido de sodio, celulosa, carbonato de magnesio, etc., vehículos inertes que tienen una funcionalidad bien establecida en la fabricación de productos farmacéuticos. Se conocen varios sistemas de administración y se pueden usar para administrar un compuesto de la presente invención que incluye la encapsulación en liposomas, micropartículas, microcápsulas y similares. Otro aspecto de la invención se refiere a una forma farmacéutica, de ahora en adelante forma farmacéutica de la invención, que comprende un compuesto de la invención (un compuesto de fórmula (I) , o la composición de la invención. En esta memoria se entiende por "forma farmacéutica" la mezcla de uno o más principios activos con o sin aditivos que presentan características físicas para su adecuada dosificación, conservación, administración y biodisponibilidad. Las formas de dosificación sólidas para administración oral pueden incluir cápsulas convencionales, cápsulas de liberación sostenida, comprimidos convencionales, comprimidos, comprimidos masticables, comprimidos sublinguales, efervescentes comprimidos, píldoras, suspensiones, polvos, gránulos y geles de liberación sostenida. En estas formas de dosificación sólidas, los compuestos activos se pueden mezclar con al menos un excipiente inerte como sacarosa, lactosa o almidón. Tales formas de dosificación también pueden comprender, como en la práctica normal, sustancias adicionales distintas de diluyentes inertes, por ejemplo, agentes lubricantes tales como estearato de magnesio. En el caso de cápsulas, comprimidos, comprimidos efervescentes y píldoras, las formas de dosificación también pueden comprender agentes tamponantes. Los comprimidos y las píldoras se pueden preparar con recubrimientos entéricos. Las formas de dosificación líquidas para administración oral pueden incluir emulsiones, soluciones, suspensiones, jarabes y elixires farmacéuticamente aceptables que contienen diluyentes inertes usados comúnmente en la técnica, tales como agua. Esas composiciones ambién pueden comprender adyuvantes tales como agentes humectantes, agentes emulsionantes y de suspensión y agentes edulcorantes, agentes aromatizantes y perfumantes. Las preparaciones inyectables, por ejemplo, suspensiones acuosas u oleaginosas, inyectables estériles se pueden formular de acuerdo con la técnica conocida usando agentes dispersantes, agentes humectantes y/o agentes de suspensión adecuados. Entre los vehículos y disolventes aceptables que se pueden utilizar se encuentran el agua, la solución de Ringer y la solución isotónica de cloruro de sodio. Los aceites estériles también se utilizan convencionalmente como disolventes o medios de suspensión. Para la administración tópica, los compuestos de la invención pueden formularse como cremas, geles, lociones, líquidos, pomadas, rocíe soluciones, dispersiones, barras sólidas, emulsiones, microemulsiones y similares que se pueden formular de acuerdo con métodos convencionales que utilizan excipientes adecuados, tales como, por ejemplo, emulsionantes, tensioactivos, agentes espesantes, agentes colorantes y combinaciones de dos o más de los mismos. Adicionalmente, el compuesto de fórmula (I) como se definió anteriormente, o una sal farmacéuticamente aceptable, estereoisómero o solvato del mismo para uso según la invención se pueden administrar en forma de parches transdérmicos o iontoforesis dispositivos. En una realización, los compuestos para uso según la invención se administran como un parche transdérmico, por ejemplo, en forma de parche transdérmico de liberación sostenida. Se conocen en la técnica parches transdérmicos adecuados. Varios sistemas de suministro de fármacos son conocidos y se pueden utilizar para administrar los agentes o composiciones para uso de acuerdo con la invención, incluyendo, por ejemplo, encapsulación en liposomas, microburbujas, emulsiones, micropartículas, microcápsulas y similares. La dosis requerida se puede administrar como una sola unidad o en una forma de liberación sostenida. Las formas de liberación sostenible y los materiales y métodos apropiados para su preparación se describen, por ejemplo, en " Modified-Release Drug Deliver y Technology ", Rathbone, MJ Hadgraft, J. y Roberts, MS (eds.) , Marcel Dekker, Inc., Nueva York (2002) , " Handbook of Pharmaceutical Controlled Release T echnology ", Wise, DL (ed.) , Marcel Dekker, Inc. Nueva York, (2000) . En una realización de la invención, la forma administrable por vía ral de un compuesto para uso según la invención está en una forma de liberación sostenida que comprende además al menos un recubrimiento o matriz. El recubrimiento o matriz de liberación sostenida incluye, sin limitación, polímeros naturales, ceras, grasas, semisintéticos o sintéticos insolubles en agua, modificados, alcoholes grasos, ácidos grasos, plastificantes naturales semisintéticos o sintéticos, o una combinación de dos o más de ellos. Los recubrimientos entéricos se pueden aplicar usando procesos convencionales conocidos por los expertos en la técnica, como se describe en, por ejemplo, Johnson, JL, " Pharmaceutical tablet coating", Coatings Technology Handbook (Segunda edición) , Satas, D. y Tracton, AA (eds. ) , Marcel Dekker, Inc. Nueva York, (2001) , Carstensen, T., " Coating Tablets in Advanced Pharmaceutical Solids", Swarbrick, J. (ed.) , Marcel Dekker, Inc. Nueva York (2001) , 455- 468. El término "prevención", "prevenir" o "prevenir", como se usa en el presente documento, se refiere a la administración de un compuesto de acuerdo con la invención o de un medicamento que comprende dicho compuesto a un sujeto que no ha sido diagnosticado como posiblemente tener una enfermedad, pero que normalmente se esperaría que la desarrolle o que tenga un mayor riesgo de padecerla. La prevención pretende evitar la aparición de dicha enfermedad. La prevención puede ser completa (por ejemplo, la ausencia total de una enfermedad) . La prevención también puede ser parcial, de modo que, por ejemplo, la aparición de una enfermedad en un sujeto es menor que la que habría ocurrido sin la administración del compuesto de la presente invención. La prevención también se refiere a la reducción de la susceptibilidad a una condición clínica. El término "tratamiento", como se usa en este documento, se refiere a cualquier tipo de terapia, que tiene como objetivo terminar, prevenir, mejorar o reducir la susceptibilidad a una condición clínica como se describe en este documento. En una realización preferida, el término tratamiento se refiere a un tratamiento profiláctico (es decir, una terapia para reducir la susceptibilidad a una afección clínica) , de un trastorno o afección como se define en el presente documento. Por tanto, "tratamiento", "tratar" y sus términos equivalentes se refieren a la obtención de un efecto farmacológico o fisiológico deseado, que cubre cualquier tratamiento de una afección o trastorno patológico en un mamífero, incluido un ser humano. El efecto puede ser profiláctico en términos de prevención total o parcial de un trastorno o síntoma del mismo y / o puede ser terapéutico en términos de una cura parcial o completa de un trastorno y / o efecto adverso atribuible al trastorno. Es decir, "tratamiento" incluye (1) evitar que el trastorno ocurra o reaparezca en un sujeto, (2) inhibir el trastorno, como detener su desarrollo, (3) detener o terminar el trastorno o, al menos, los síntomas asociados con el mismo, de modo que el huésped ya no sufra el trastorno o sus síntomas, como provocar la regresión del trastorno o sus síntomas, por ejemplo, al restaurar o reparar una función perdida, altante o defectuosa, o estimular un proceso ineficiente, o (4) aliviar, aliviar o mejorar el trastorno o los síntomas asociados con el mismo. El término "sujeto" como se usa en el presente documento, se refiere a cualquier sujeto, particularmente un sujeto mamífero, para el que se desea la terapia. Los sujetos mamíferos incluyen humanos, animales domésticos, animales de granja y zoológicos, deportes o animales de compañía como perros, gatos, cobayas, conejos, ratas, ratones, caballos, ganado, vacas, etc. En una realización preferida de la invención, el sujeto es un mamífero. En una realización más preferida de la invención, el sujeto es un ser humano. La administración de los compuestos, composiciones o formas farmacéuticas de la presente invención puede ser realizada mediante cualquier método adecuado, como la infusión intravenosa y las vías oral, tópica o parenteral. La administración oral es la preferida por la conveniencia de los pacientes. La cantidad administrada de un compuesto de la presente invención dependerá de la relativa eficacia del compuesto elegido, la severidad de la enfermedad a tratar y el peso del paciente. Sin embargo, los compuestos de esta invención serán administrados una o más veces al día, por ejemplo 1, 2, 3 ó 4 veces diarias, con una dosis total entre 0.1 y 1000 mg/Kg/día. Es importante tener en cuenta que puede ser necesario introducir variaciones en la dosis, dependiendo de la edad y de la condición del paciente, así como modificaciones en la vía de administración. A lo largo de la descripción y las reivindicaciones la palabra "comprende" y sus variantes no pretenden excluir otras características técnicas, aditivos, componentes o pasos. Para los expertos en la materia, otros objetos, ventajas y características de la invención se desprenderán en parte de la descripción y en parte de la práctica de la invención. Los siguientes ejemplos y figuras se proporcionan a modo de ilustración, y no se pretende que sean limitativos de la presente invención. EJEMPLOS DE LA INVENCIÓN Cribado virtual Con el objetivo de encontrar nuevos inhibidores para la hepsina se realizó un cribado virtual mediante la técnica de docking molecular a partir de la estructura cristalográfica de la proteína epositada en la Protein Data Bank con código 1P57 frente a la librería de compuestos DrugBank (https://go.drugbank.com/) en las coordenadas de su sitio catalítico, caracterizada por residuos HIS57, ASP102 y SER195. El docking molecular se realizó en un clúster de cómputo de alto rendimiento mediante el software Autodock Vina 1.1.2 (http://vina.scripps.edu/) y para ello se convirtieron tanto la estructura de la proteína como la librería de ligandos DrugBank al formato pdbqt mediante MGLTools. Una vez terminados los cálculos se priorizaron los compuestos en función de docking score y se examinaron visualmente los 8 primeros, seleccionando el compuesto Suramina (código DrugBank DB04786 y primero en la lista) con un docking score de -12 Kcal/mol e interaciones hidrofóbicas con los residuos ALA-39, LEU-41, HIS-57, PRO-60, PHE-97, GLN-151, interacciónes mediante puente de hidrogeno con los residuos GLY-38, HIS-40, HIS-57, ASN-63, GLN-73, ASN-99, GLN-151, GLY-153, SER-195, SER-214, GLY-216, GLY-219, interacciones por "puentes de sal" con ARG-34, HIS-57 y una interacción "piStacking" con el residuo HIS-57 como se aprecia en la Figura 1. En dicha figura podemos observar que Suramina impide el acceso a la triada catalítica, representada en formato de superficie para sus tres residuos Suramina actúa como inhibidor irreversible de hepsina. Mediante un ensayo fluorogénico se determinó la capacidad de Suramina de inhibir la actividad proteolítica de hepsina. Hepsina es una serín proteasas capaz de hidrolizar al sustrato BOC-Gln-Arg-Arg-AMC (Bachem, Barcelona, Spain) , de manera que es posible registrar la emisión de fluorescencia mediante un lector de placas con longitudes de onda de excitación y emisión de 380 nm y 460 nm, respectivamente. Para ello, se incubó hepsina (0.05 M) (R&D Systems, Madrid, Spain) en tampón 50 mM Tris-HCl, pH 9 buffer, con 200 M BOC-Gln-Arg-Arg-AMC y se registró la fluorescencia emitida durante 5 minutos. Para comprobar el efecto de Suramina, se llevó a cabo la misma reacción, pero incubando previamente hepsina con Suramina durante 1 hora a 37°C y en distintas concentraciones que oscilaron entre 0.13 y 10 M. De esta forma se calculó el valor de IC50, o concentración de inhibidor a la que se alcanza la mitad de la velocidad máxima de hepsina en la hidrólisis de su sustrato, que fue de 0.66 M. Suramina reduce la migración de células de cáncer colorrectal Una vez que las células alcanzan confluencia, forman una monocapa. Mediante la eliminación de una tira de células 300-500 m de anchura con una punta de pipetea de 200 l, es posible evaluar la capacidad de migración de las células y el efecto de Suramina en este proceso. La capacidad de migración se evalúa en función del porcentaje de área ocupada tras 48 horas de incubación y se cuantifica utilizando el software ImageJ. Los resultados muestran que Suramina a una concentración de 0, 66 M reduce de forma significativa la migración de células Caco-2, tanto con expresión basal como con sobreexpresión de hepsina, mediante transfección estable de un plásmido que contiene el gen HPN. Suramina reduce la invasión de células de cáncer colorrectal Para evaluar la invasión se realizó un ensayo en el que se evaluó la capacidad de las células para degradar una matriz de gelatina. Para ello, se mezcló gelatina al 0.2% y rodamina (Invitrogen, Life Technologies, Madrid, Spain) , en una ratio 1:55 en tampón NaCl 61 mM, borohidrato sódico 50 mM y después se dializó toda la noche en PBS. Se prepararon cubres con la mezcla preparada cubriéndolos y se fijó la matriz con glutaraldehido 0.5% durante 15 min. Los cubres se lavaron con PBS y se añadieron las suspensiones de células Caco-2 durante 72 horas. Posteriormente se fijaron las células con formaldehído 3.7% y se incubó con phalloidin faloidin?? (0, 01 mg/ml) y ProLongGold Antifade con DAPI (4', 6-diamino-2-phenylindole; ThermoFisher) . Las imágenes se tomaron con un microscopio confocal SP8 LEICA y se analizaron con ImageJ. Los resultados mostraron que Suramina, a una concentración de 0, 66 M, reduce de forma significativa la degradación de la gelatina de células Caco-2 tanto con expresión basal como con sobreexpresión de hepsina. Suramina reduce la generación de trombina Puesto que hepsina activa al FVII y, por tanto, la cascada de la coagulación, podría contribuir al estado de hipercoagulabilidad de los pacientes con cáncer colorrectal con niveles elevados de hepsina. Para comprobar la hipótesis, se incubó plasma procedente de 20 sujetos sanos, a los que se extrajo sangre citratada 3.8% mediante venopunción, con las células Caco-2 con sobreexpresión de hepsina. El plasma se centrifugó previamente a 2500 g 20 minutos. Para el ensayo de generación de trombina, se retiró el plasma incubado y se incubó con el reactivo LOW® (factor tisular: 1 pmol; fosfolípidos: 4 pmol; Diagnostica Stago) en una placa de 96 pocillo. Todas las muestras se prepararon en duplicado. La coagulación en estas muestras se inició añadiendo cloruro cálcico en un tampón que contenía el sustrato fluorogénico FluCa-kit reagent® (Diagnostica Stago) . Para cada muestra individual se utilizó un calibrador de rombina. La fluorescencia se registró durante 60 min en un fluorímetro Fluoroskan Ascent (Thermolab Systems) y los datos se analizaron utilizando el software Thrombinoscope™ (version 5.0.0.742; Diagnostica Stago) . Se analizaron los siguientes parámetros: (a) lag-time, que indica el inicio de la fase de generación de trombina; (b) tiempo hasta alcanzar la máxima concentración de trombina (ttPeak) ; (c) concentración máxima de trombina (Peak) ; (d) índice de frecuencia media (MRI) de la fase de propagación de la generación de trombina calculada por la fórmula Peak/ (ttPeak - lag-time) y se expresa en nM/min; y (e) potencial endógeno de trombina (ETP) que muestra la actividad enzimática de trombina evaluada como el área bajo la curva. Los resultados muestran que Suramina reduce el potencial endógeno de trombina (ETP) , el pico máximo (peak) y la velocidad en alcanzar el pico mientras que provoca un incremento en el tiempo de inicio de la fase de generación de trombina (lag-time) y el tiempo en alcanzar el pico. Tabla 1. Valores promedios del test de generación de trombina. Suramina no afecta ni a la muerte celular, ni a la proliferación celular Se quiso evaluar si la menor migración o invasión celular podría deberse a una mayor muerte, o a una menor proliferación celular. Para ello se incubaron las células (5x105 cells/ml) en placas de 6 pocillos en presencia o ausencia de Suramina tras 48 h. El tipo de muerte celular se determinó mediante el ensayo de anexina V/7-ADD (Anexin V Apoptosis Detection Kit FITC; eBiosciences, Thermo Fisher, Karlsruhe, Alemania; 7-ADD BD-Biosciences) siguiendo las instrucciones del fabricante. Para evaluar la actividad proliferativa de las células Caco-2, se añadió 5-etinil-2'-desoxiuridina (EdU) (Thermo Fisher Scientific) durante 48 h. Luego, se usaron alícuotas de las células tumorales para determinar el porcentaje de células positivas, es decir, Caco-2 o Caco-2-HPN-EdU+, detectadas por reacción de acoplamiento de azida luorescente con EdU de acuerdo con el protocolo del fabricante (Click-iT; Thermo Fisher Scientific) . Los resultados mostraron que Suramina no afecta ni a la muerte (porcentajes entorno al 5-10 %) ni a la proliferación celular. Suramina inhibe la invasión de las células de cáncer colorrectal en modelos de xenotrasplante de larvas de pez cebra Se obtuvieron peces cebra wild type (Danio rerio H. Cypriniformes, Cyprinidae) del Zebrafish International Resource Center (ZIRC, Oregon, EE. UU.) Los peces roya9/a9; nacrew2/w2 (casper) transparentes se obtuvieron como ya se ha descrito previamente en la bibliografía [R.M. White, A. Sessa, C. Burke, T. Bowman, J. LeBlanc, C. Ceol, et al. Transparent adult zebrafish as a tool for in vivo transplantation analysis. Cell Stem Cell 2008;2:183-9]. Los peces se aparearon, clasificaron, criaron y procesaron como se describe en el manual del pez cebra [M. Westertield. The zebrafish book: a guide for the laborator y use of zebrafish. Http//Zfin Org/Zf_info/Zfbook/Zfbk Html 2000.]. Los huevos de pez cebra fertilizados se obtuvieron del desove natural y se mantuvieron en nuestras instalaciones siguiendo las prácticas estándar de cría. Los animales se mantuvieron en un ciclo de luz / oscuridad de 12 h a 28 ° C. Las larvas de pez cebra se anestesiaron con 0.16 mg/ml ethyl 3-aminobenzoato (tricaine; Sigma-Aldrich) . Las células Caco-2 y Caco-2-HPN se cultivaron en presencia y ausencia de 0, 66 M de suramina durante 48 h, luego se disgregaron y marcaron con 1, 1'-di-octa-decil-3, 3, 3 ', 3'-perclorato de tetra-metil-indo-carbo-cianina (Dil, ThermoFisher) y finalmente se resuspendieron en un tampón que contenía suero bovino fetal al 5% en PBS. La inyección de las células en embriones se realizó mediante la inyección de 200 células/embrión en el saco vitelino de larvas de pez cebra wild type o Casper 48 horas después de la fertilización y después de 5 días a 35°C, las larvas se analizaron mediante microscopía de fluorescencia para comprobar la diseminación de las células tumorales. La puntuación de la invasión de células Caco-2 y Caco-2-HPN se calculó como el porcentaje de larvas con invasión de células tumorales con respecto al total de larvas analizadas. El pretratamiento de las células Caco-2-HPN (CACO HPN) con suramina (HPN + S) redujo su capacidad de invasión a los niveles encontrados en las células parentales (CACO WT) (Figura 8) , lo que confirma los estudios in vitro y muestra además su potencial terapéutico para el tratamiento del cáncer colorrectal.
+ ES-2912350_A1 Nuevo tratamiento del cáncer colorrectal CAMPO DE LA INVENCIÓN La presente invención pertenece al campo de la biomedicina, y más concretamente se refiere a un nuevo tratamiento para el cáncer colorrectal. ANTECEDENTES DE LA INVENCIÓN El cáncer colorrectal es el tercer cáncer más común diagnosticado tanto en hombres como en mujeres. Tiene lugar en el colon y recto y se desarrolla lentamente a partir de pólipos adenomatosos. El crecimiento excesivo de la mucosa del colon genera estos pólipos que tienen una morfología pedunculada o sésil. El desarrollo de cáncer colorrectal puede tener lugar por diferentes vías. Entre ellas se encuentra la aparición de lesiones serradas con apariencia del epitelio en dientes de sierra y que ocurre en el colon proximal. Esta vía se detecta en un tercio de todos los cánceres colorrectales. Con respecto a la secuencia adenoma-carcinoma, se ha propuesto que la progresión desde adenomas pequeños a grades adenomas o adenocarcinomas avanzados está mediada por múltiples rutas moleculares que incluyen la inestabilidad de microsatélites, la inestabilidad cromosómica y rutas epigenéticas (Jebelli, Asiyeh et al. Medicinal research reviews vol. 41, 1 (2021) : 395-434) . Es posible hacer una prevención precoz puesto que se ha estimado que al menos lleva 10 años que un pólipo se transforme en célula tumoral (0ines, Mari et al. "Epidemiology and risk factors of colorectal polyps." Best practice & research. Clinical gastroenterology vol. 31, 4 (2017) : 419-424) . El diagnóstico precoz mediante colonoscopia y detección de sangre oculta en heces, así como la mejora en los tratamientos ha contribuido a prolongar la supervivencia de los tumores colorrectales en la fase de curación, Sin embargo, la metástasis está presente en aproximadamente el 25% de los pacientes durante el diagnóstico y en total, el 50% de los pacientes con cáncer colorrectal desarrollará metástasis (Ohhara, Yoshihito et al. World journal of gastrointestinal oncology vol. 8, 9 (2016) : 642-55) . Debido a la heterogeneidad y naturaleza multifactorial del cáncer colorrectal han aumentado considerablemente en los últimos años estudios para identificar predictores clínicos y moleculares así como factores pronósticos con potencial para mejorar el manejo de los pacientes (Aran, Veronica et al. Clinical colorectal cancer vol. 15, 3 (2016) : 195-203) . Tanto la incidencia como la mortalidad del cáncer colorrectal ha disminuido tanto para hombres como para mujeres desde 1970 debido a la detección precoz. Sin embargo, aunque diferentes terapias estás disponibles para el cáncer colorrectal metastásico, los resultados no son óptimos debido a la heterogeneidad entre pacientes debido a características moleculares, respuesta a la terapia y presentación clínica (Linnekamp, Janneke F et al. Cancer research vol. 75, 2 (2015) : 245-9) . Los tratamientos más comunes de estos tumores se basan en la cirugía, la quimioterapia, terapias dirigidas y el tratamiento hormonal sustitutivo (Rejhová, A et al. European journal of medicinal chemistr y vol. 144 (2018) : 582-594) . Al menos el 50% de los pacientes con cáncer colorrectal recaen tras la resección quirúrgica y finalmente mueren de enfermedad metastásica. A pesar de la quimioterapia adyuvante postoperatoria en estos pacientes para disminuir el riesgo de recurrencia, el tratamiento adyuvante no da beneficios en supervivencia, especialmente en pacientes diagnosticados con estadío I (Aran, Veronica et al. Clinical colorectal cancer vol. 15, 3 (2016) : 195-203) . La terapia dirigida está basada en anticuerpos monoclonales y pequeños inhibidores moleculares, que selectivamente bloquean la proliferación celular mediante la intervención con ciertas moléculas y proteínas sobreexpresadas requeridas para la expansión del tumor (Jebelli, Asiyeh et al. Medicinal research reviews vol. 41, 1 (2021) : 395-434) . Las proteasas pericelulares han sido ampliamente implicadas en carcinogénesis, no solo porque actúan como enzimas capaces de degradar la matriz extracelular, permitiendo así a las células tumorales romper la membrana basal e invadir el tejido circundante, sino que son capaces de actuar como modificadores proteolíticos de factores de crecimiento y receptores activados por proteasas que son críticos para la activación de rutas de señalización tumoral (Tanabe, Lauren M, and Karin List. The FEBS journal vol. 284, 10 (2017) : 1421-1436) . Entre estas proteasas se encuentra hepsina, que pertenece a la super familia de serín proteasas transmembrana de tipo II (TTSP) . La Hepsina se encuentra sobreexpresada en cáncer de próstata y sus niveles elevados son indicativos de mal pronóstico y recaída tras prostatectomía radical (Sardana, Girish et al. Clinical chemistr y vol. 54, 12 (2008) : 1951-60.) . También se ha demostrado su sobreexpresión en cáncer de ovario (Miao, Jiangyong et al. International journal of cancer vol. 123, 9 (2008) : 2041-7) y cáncer de mama (Miao, Jiangyong et al. International journal of cancer vol. 123, 9 (2008) : 2041-7) . El desarrollo de eventos tromboembólicos venosos (ETEV) es un problema común en los tumores digestivos. La incidencia acumulada de ETEV en series modernas varía entre 12 20% (Posch, Florian et al. Thrombosis and haemostasis vol. 115, 4 (2016) : 817-26) , dependiendo de las disparidades en las características de los pacientes en cada estudio y el contexto en el que la evaluación es llevada a cabo. La trombogénesis se atribuye a una combinación de factores clínicos y biológicos, que incluyen la activación del sistema hemostático por parte del tumor y la toxicidad vascular de la quimioterapia. La trombosis se asocia con una mayor morbilidad y mortalidad en estos pacientes (Carmona-Bayonas, A et al. British journal of cancer vol. 116, 8 (2017) : 994-1001) y es uno de los eventos adversos más importantes en los ensayos clínicos de terapia anti-neoplásica. BREVE DESCRIPCIÓN DE LOS DIBUJOS Fig. 1. Fórmula de Suramín. Fig. 2. Pose resultante del docking molecular entre Suramín y hepsina. Fig. 3. Cálculo del valor de IC50 de Suramín sobre la actividad de Hepsina. Velocidad máxima de la actividad de Hepsina en presencia de dosis crecientes de Suramín. Fig. 4. Efecto de Suramín en la migración de células de cáncer colorrectal. Porcentaje de migración celular de las células Caco-2 y Caco-2-HPN en presencia o ausencia de Suramín. Fig. 5. Efecto de Suramín en la degración de gelatina de células de cáncer colorrectal. Porcentaje de degradación celular de las células Caco-2 y Caco-2-HPN en presencia o ausencia de Suramín. Fig. 6. Efecto de Suramín en la generación de trombina. Valores del potencial endógeno de trombina en células Caco-2 y Caco-2-HPN. Valores del tiempo hasta el inicio del pico (Lagtime) , yel tiempo en alcanzar el pico máximo (ttPeak) en las células Caco-2-HPN en presencia o ausencia de Suramín. Fig. 7. Efecto de Suramín en la proliferación de células de cáncer colorrectal. Porcentaje de células Caco-2 y Caco-2-HPN que están proliferando (EdU+) en presencia o ausencia de Suramín. DESCRIPCIÓN DE LA INVENCIÓN Los autores de la presente invención han demostrado que el Suramin reduce la migración de células de cáncer colorrectal, reduce la invasión de células de cáncer colorrectal, reduce la generación de trombina en plasma procedente de sujetos sanos tras la incubación con células de cáncer colorrectal y reduce la generación de trombina en plasma de pacientes con cáncer colorrectal con elevados niveles de hepsina en plasma. Han demostrado que el suramin, un fármaco utilizado para el tratamiento de la tripanosomiasis africana, la oncocercosis y en investigación para el tratamiento del cáncer de próstata (PMID: 15484217) , es inhibidor irreversible de hepsina. Por tanto, un primer aspecto de la invención se refiere al un compuesto de fórmula (I) Fórmula (I) o cualquiera de sus sales, preferiblemente cualquier sal farmacéuticamente aceptable, ésteres, tautómeros, polimorfos, hidratos farmacéuticamente aceptables, o un isómero, profármacos, derivados, solvatos o análogos, o cualquiera de sus combinaciones, para la prevención o tratamiento del cáncer colorrectal. Como se puede observar en la Fig. 4, el Suramin es capaz de disminuir la migración de las células del cáncer colorrectal. Además, reduce de forma significativa la degradación de la gelatina de células Caco-2 tanto con expresión basal como con sobreexpresión de hepsina (Fig. 5) . Por tanto, en una realización preferida de este aspecto de la invención, el cáncer colorrectal es el cáncer colorrectal metastásico. Más preferiblemente, donde el individuo presenta los niveles de hepsina elevados con respecto a los niveles normales. Tal y como se muestra en la Fig. 6, el compuesto de la invención, reduce la generación de trombina en plasma de pacientes con cáncer colorrectal con elevados niveles de hepsina en plasma. Por tanto, otro aspecto de la invención se refiere al compuesto de la invención para la prevención o tratamiento de los eventos tromboembólicos venosos (ETEV) , y preferiblemente de aquellos desarrollados como consecuencia de la terapia antineoplásica. Los compuestos de fórmula (I) , o sus sales o solvatos para uso de acuerdo con la invención están preferiblemente en forma farmacéuticamente aceptable o sustancialmente pura. Por forma farmacéuticamente aceptable se entiende, entre otras cosas, que tiene un nivel de pureza farmacéuticamente aceptable excluyendo los aditivos farmacéuticos normales tales como diluyentes y vehículos, y que no incluye ningún material considerado tóxico a niveles de dosificación normales. Los niveles de pureza de la sustancia farmacéutica están preferiblemente por encima del 50%, más preferiblemente por encima del 70%, lo más preferiblemente por encima del 90%. En una realización preferida, está por encima del 95% del compuesto de fórmula (I) o de su sal, estereoisómero o solvato farmacéuticamente aceptable. La invención también se refiere a usos según la invención de metabolitos de los compuestos descritos en la presente descripción. Un "metabolito" de un compuesto descrito en el presente documento es un derivado de ese compuesto que se forma cuando el compuesto se metaboliza. El término "metabolito activo" se refiere a un derivado biológicamente activo de un compuesto que se forma cuando el compuesto se metaboliza. El término "metabolizado", como se usa en este documento, se refiere a la suma de los procesos (que incluyen, pero no se limitan a, reacciones de hidrólisis y reacciones catalizadas por enzimas) mediante los cuales un organismo cambia una sustancia particular. Por tanto, las enzimas pueden producir alteraciones estructurales específicas en un compuesto. Después de entrar en el cuerpo, la mayoría de los medicamentos son sustratos para reacciones químicas que pueden cambiar sus propiedades físicas y efectos biológicos. Estas conversiones metabólicas, que suelen afectar a la polaridad de los compuestos de la invención, alteran la forma en que los fármacos e distribuyen y excretan en el organismo. Sin embargo, en algunos casos, se requiere el metabolismo de un fármaco para obtener un efecto terapéutico. La invención también se refiere a profármacos de los compuestos para uso según la invención. El término "prodroga" o "profármaco" tal como aquí se utiliza incluye cualquier derivado de un compuesto de fórmula (I) , -por ejemplo y no limitativamente: ésteres (incluyendo ésteres de ácidos carboxílicos, ésteres de aminoácidos, ésteres de fosfato, ésteres de sulfonato de sales metálicas, etc.) , carbamatos, amidas, etc.- que al ser administrado a un individuo puede ser transformado directa o indirectamente en dicho compuesto de fórmula (I) en el mencionado individuo. Ventajosamente, dicho derivado es un compuesto que aumenta la biodisponibilidad del compuesto de fórmula (I) cuando se administra a un individuo o que potencia la liberación del compuesto de fórmula (I) en un compartimento biológico. La naturaleza de dicho derivado no es crítica siempre y cuando pueda ser administrado a un individuo y proporcione el compuesto de fórmula (I) , en un compartimento biológico de un individuo. La preparación de dicho profármaco puede llevarse a cabo mediante métodos convencionales conocidos por los expertos en la materia, que son capaces de liberar el compuesto de fórmula (I) como ingrediente activo cuando el profármaco se administra a un sujeto mamífero. La liberación del ingrediente activo ocurre in vivo. Los profármacos se pueden preparar mediante técnicas conocidas por los expertos en la técnica. Estas técnicas generalmente modifican los grupos funcionales apropiados en un compuesto dado. Sin embargo, estos grupos funcionales modificados regeneran grupos funcionales originales mediante manipulación rutinaria o in vivo. Los profármacos de los compuestos de la invención incluyen compuestos en los que se modifica un grupo hidroxi, amino, carboxílico o similar. Los ejemplos de profármacos incluyen, pero no se limitan a, ésteres. Los compuestos de fórmula general (I) y sus profármacos que contienen uno o varios centros quirales pueden estar presentes como racematos, mezclas diastereoméricas o isómeros individuales ópticamente activos. Los compuestos de la presente invención representados por la fórmula (I) pueden incluir isómeros, dependiendo de la presencia de enlaces múltiples, incluyendo isómeros ópticos o enantiómeros, dependiendo de la presencia de centros quirales. Los isómeros, enantiómeros o diastereoisómeros individuales y las mezclas de los mismos caen dentro del alcance de la presente invención, es decir, el término isómero también se refiere a cualquier mezcla de isómeros, como diastereómeros, racémicos, etc., incluso a sus isómeros ópticamente activos o las mezclas en distintas proporciones de los mismos. Los enantiómeros o diastereoisómeros individuales, así como sus mezclas, pueden separarse mediante técnicas convencionales. Composiciones y composiciones farmacéuticas El compuesto de fórmula (I) de la invención puede estar formando parte de una composición, preferiblemente de una composición farmacéutica. Otro aspecto de la invención se refiere a una composición que comprende el compuesto de fórmula (I) de la invención, de ahora en adelante composición de la invención, o una sal, estereoisómero, tautómero, hidrato o solvato farmacéuticamente aceptable del mismo, para la prevención y tratamiento del cáncer colorrectal. En una realización preferida de este aspecto de la invención, el cáncer colorrectal es el cáncer colorrectal metastásico. Más preferiblemente, donde el individuo presenta los niveles de hepsina elevados con respecto a los niveles normales. Otro aspecto de la invención se refiere al compuesto de la invención para la prevención o tratamiento de los eventos tromboembólicos venosos (ETEV) , y preferiblemente de aquellos desarrollados como consecuencia de la terapia antineoplásica. En otra realización de este aspecto, la composición de la invención además comprende otro principio activo. Más preferiblemente es una composición farmacéutica. En otra realización preferida además comprende vehículos farmacéuticamente aceptables. En otra realización preferida de este aspecto, la primera composición de la invención solo comprende como principio activo el compuesto de fórmula (I) , aunque puede comprender, además, excipientes y vehículos farmacéuticamente aceptables. El término "medicamento", tal y como se usa en esta memoria, hace referencia a cualquier sustancia usada para prevención, diagnóstico, alivio, tratamiento o curación de enfermedades en el hombre y los animales. La administración de los compuestos, composiciones o formas farmacéuticas de la presente invención puede ser realizada mediante cualquier método adecuado, como la infusión intravenosa y las vías oral, tópica o parenteral. La administración oral es la preferida por la conveniencia de los pacientes. La cantidad administrada de un compuesto de la presente invención dependerá de la relativa eficacia del compuesto elegido, la severidad de la enfermedad a tratar y el peso del paciente. Sin embargo, los compuestos de esta invención serán administrados una o más veces al día, por ejemplo 1, 2, 3 O 4 veces diarias, con una dosis total entre 0.1 y 1000 mg/Kg/día. Es importante tener en cuenta que puede ser necesario introducir variaciones en la dosis, dependiendo de la edad y de la condición del paciente, así como modificaciones en la vía de administración. "Composición farmacéutica", como se usa en el presente documento, se refiere a composiciones y entidades moleculares que son fisiológicamente tolerables y no producen típicamente una reacción alérgica o una reacción desfavorable similar a como trastornos gástricos, mareos y similares, cuando se administra a un humano o animal. Preferiblemente, el término "farmacéuticamente aceptable" significa que está aprobado por una agencia reguladora de un gobierno estatal o federal o está incluido en la Farmacopea de los Estados Unidos u otra farmacopea generalmente reconocida para uso en animales, y más particularmente en humanos. El término "excipiente" se refiere a un vehículo, diluyente o adyuvante que se administra con el ingrediente activo. Dichos excipientes farmacéuticos pueden ser líquidos estériles, como agua y aceites, incluidos los de origen petrolífero, animal, vegetal o sintético, como aceite de cacahuete, aceite de soja, aceite mineral, aceite de sésamo y similares. Como vehículos se utilizan preferiblemente agua o soluciones acuosas salinas y soluciones acuosas de dextrosa y glicerol, particularmente para soluciones inyectables. Los vehículos farmacéuticos adecuados se describen en "Remington's Phamaceutical Sciences" de EW Martin, 21a edición, 2005; o "Manual de excipientes farmacéuticos", Rowe CR; Paul JS; Marian EQ, sexta edición. Como se emplea aquí, el término "principio activo", "substancia activa", "substancia farmacéuticamente activa", "ingrediente activo" ó "ingrediente farmacéuticamente activo" significa cualquier componente que potencialmente proporcione una actividad farmacológica u otro efecto diferente en el diagnóstico, cura, mitigación, tratamiento, o prevención de una enfermedad, o que afecta a la estructura o función del cuerpo del hombre u otros animales. El término incluye aquellos componentes que promueven un cambio químico en la elaboración del fármaco y están presentes en el mismo de una forma modificada prevista que proporciona la actividad específica o el efecto. Las cantidades apropiadas de un compuesto de fórmula (I) como se define anteriormente, o una sal, estereoisómero farmacéuticamente aceptable o solvato del mismo se pueden formular con excipientes y/o vehículos farmacéuticamente aceptables para obtener una composición farmacéutica para uso en los usos médicos de la invención. Los vehículos farmacéuticamente aceptables adecuados incluyen, por ejemplo, agua, soluciones salinas, alcohol, aceites vegetales, polietilenglicoles, gelatina, lactosa, amilosa, estearato de magnesio, talco, tensioactivos, ácido silícico, parafina viscosa, aceite perfumado, monoglicéridos y diglicéridos. de ácidos grasos, ésteres de ácidos grasos, petroetrales, hidroximetilcelulosa, polivinilpirrolidona y similares. Las composiciones farmacéuticas que contienen el compuesto de fórmula (I) , como se definió anteriormente, o una sal, estereoisómero o solvato farmacéuticamente aceptable del mismo para su uso de acuerdo con la invención pueden ocurrir en cualquier forma farmacéutica de administración considerada apropiada para la vía de administración seleccionada, por ejemplo, por administración sistémica (por ejemplo, inyección intravenosa, subcutánea, intramuscular) , oral, parenteral o tópica, para lo cual incluirá los excipientes farmacéuticamente aceptables necesarios para la formulación del método de administración deseado. Además, también es posible administrar la composición que comprende el compuesto de fórmula (I) como se definió anteriormente, o una sal farmacéuticamente aceptable, estereoisómero o solvato del mismo para uso según la invención por vía intranasal o sublingual que permite la administración sistémica mediante un modo de administración no agresivo. Además, la administración intraventricular puede ser adecuada. Una vía de administración preferida es la oral. Cuando sea necesario, el compuesto de fórmula (I) como se definió anteriormente, o una sal, estereoisómero o solvato farmacéuticamente aceptable del mismo para su uso de acuerdo con la invención está comprendido en una composición que también incluye un agente solubilizante y un anestésico local para aliviar cualquier dolor en el lugar de la inyección. Generalmente, los ingredientes se suministran por separado o mezclados en forma de dosificación unitaria, por ejemplo, como un polvo liofilizado seco o un concentrado libre de agua en un recipiente herméticamente cerrado como una ampolla o bolsita que indica la cantidad de agente activo. Cuando la composición deba administrarse mediante infusión, se puede dispensar con una botella de infusión que contenga agua o solución salina estériles de calidad farmacéutica. En casos distintos de la administración intravenosa, la composición puede contener cantidades menores de agentes humectantes o emulsionantes, o agentes tamponantes del pH. La composición puede ser una solución líquida, suspensión, emulsión, gel, polímero o formulación de liberación sostenida. La composición se puede formular con aglutinantes y vehículos tradicionales, como se conoce en la técnica. Las formulaciones pueden incluir vehículos estándar tales como grados farmacéuticos de manitol, lactosa, almidón, estearato de magnesio, sacárido de sodio, celulosa, carbonato de magnesio, etc., vehículos inertes que tienen una funcionalidad bien establecida en la fabricación de productos farmacéuticos. Se conocen varios sistemas de administración y se pueden usar para administrar un compuesto de la presente invención que incluye la encapsulación en liposomas, micropartículas, microcápsulas y similares. Otro aspecto de la invención se refiere a una forma farmacéutica, de ahora en adelante forma farmacéutica de la invención, que comprende un compuesto de la invención (un compuesto de fórmula (I) , o la composición de la invención. En esta memoria se entiende por "forma farmacéutica" la mezcla de uno o más principios activos con o sin aditivos que presentan características físicas para su adecuada dosificación, conservación, administración y biodisponibilidad. Las formas de dosificación sólidas para administración oral pueden incluir cápsulas convencionales, cápsulas de liberación sostenida, comprimidos convencionales, comprimidos, comprimidos masticables, comprimidos sublinguales, efervescentes comprimidos, píldoras, suspensiones, polvos, gránulos y geles de liberación sostenida. En estas formas de dosificación sólidas, los compuestos activos se pueden mezclar con al menos un excipiente inerte como sacarosa, lactosa o almidón. Tales formas de dosificación también pueden comprender, como en la práctica normal, sustancias adicionales distintas de diluyentes inertes, por ejemplo, agentes lubricantes tales como estearato de magnesio. En el caso de cápsulas, comprimidos, comprimidos efervescentes y píldoras, las formas de dosificación también pueden comprender agentes tamponantes. Los comprimidos y las píldoras se pueden preparar con recubrimientos entéricos. Las formas de dosificación líquidas para administración oral pueden incluir emulsiones, soluciones, suspensiones, jarabes y elixires farmacéuticamente aceptables que contienen diluyentes inertes usados comúnmente en la técnica, tales como agua. Esas composiciones también pueden comprender adyuvantes tales como agentes humectantes, agentes mulsionantes y de suspensión y agentes edulcorantes, agentes aromatizantes y perfumantes. Las preparaciones inyectables, por ejemplo, suspensiones acuosas u oleaginosas, inyectables estériles se pueden formular de acuerdo con la técnica conocida usando agentes dispersantes, agentes humectantes y/o agentes de suspensión adecuados. Entre los vehículos y disolventes aceptables que se pueden utilizar se encuentran el agua, la solución de Ringer y la solución isotónica de cloruro de sodio. Los aceites estériles también se utilizan convencionalmente como disolventes o medios de suspensión. Para la administración tópica, los compuestos de la invención pueden formularse como cremas, geles, lociones, líquidos, pomadas, rocíe soluciones, dispersiones, barras sólidas, emulsiones, microemulsiones y similares que se pueden formular de acuerdo con métodos convencionales que utilizan excipientes adecuados, tales como, por ejemplo, emulsionantes, tensioactivos, agentes espesantes, agentes colorantes y combinaciones de dos o más de los mismos. Adicionalmente, el compuesto de fórmula (I) como se definió anteriormente, o una sal farmacéuticamente aceptable, estereoisómero o solvato del mismo para uso según la invención se pueden administrar en forma de parches transdérmicos o iontoforesis dispositivos. En una realización, los compuestos para uso según la invención se administran como un parche transdérmico, por ejemplo, en forma de parche transdérmico de liberación sostenida. Se conocen en la técnica parches transdérmicos adecuados. Varios sistemas de suministro de fármacos son conocidos y se pueden utilizar para administrar los agentes o composiciones para uso de acuerdo con la invención, incluyendo, por ejemplo, encapsulación en liposomas, microburbujas, emulsiones, micropartículas, microcápsulas y similares. La dosis requerida se puede administrar como una sola unidad o en una forma de liberación sostenida. Las formas de liberación sostenible y los materiales y métodos apropiados para su preparación se describen, por ejemplo, en " Modified-Release Drug Deliver y Technology ", Rathbone, MJ Hadgraft, J. y Roberts, MS (eds.) , Marcel Dekker, Inc., Nueva York (2002) , " Handbook of Pharmaceutical Controlled Release Technology ", Wise, DL (ed.) , Marcel Dekker, Inc. Nueva York, (2000) . En una realización de la invención, la forma administrable por vía oral de un compuesto para uso según la invención está en una forma de liberación sostenida ue comprende además al menos un recubrimiento o matriz. El recubrimiento o matriz de liberación sostenida incluye, sin limitación, polímeros naturales, ceras, grasas, semisintéticos o sintéticos insolubles en agua, modificados, alcoholes grasos, ácidos grasos, plastificantes naturales semisintéticos o sintéticos, o una combinación de dos o más de ellos. Los recubrimientos entéricos se pueden aplicar usando procesos convencionales conocidos por los expertos en la técnica, como se describe en, por ejemplo, Johnson, JL, " Pharmaceutical tablet coating", Coatings Technology Handbook (Segunda edición) , Satas, D. y Tracton, AA (eds. ) , Marcel Dekker, Inc. Nueva York, (2001) , Carstensen, T., " Coating Tablets in Advanced Pharmaceutical Solids", Swarbrick, J. (ed.) , Marcel Dekker, Inc. Nueva York (2001) , 455-468. El término "prevención", "prevenir" o "prevenir", como se usa en el presente documento, se refiere a la administración de un compuesto de acuerdo con la invención o de un medicamento que comprende dicho compuesto a un sujeto que no ha sido diagnosticado como posiblemente tener una enfermedad, pero que normalmente se esperaría que la desarrolle o que tenga un mayor riesgo de padecerla. La prevención pretende evitar la aparición de dicha enfermedad. La prevención puede ser completa (por ejemplo, la ausencia total de una enfermedad) . La prevención también puede ser parcial, de modo que, por ejemplo, la aparición de una enfermedad en un sujeto es menor que la que habría ocurrido sin la administración del compuesto de la presente invención. La prevención también se refiere a la reducción de la susceptibilidad a una condición clínica. El término "tratamiento", como se usa en este documento, se refiere a cualquier tipo de terapia, que tiene como objetivo terminar, prevenir, mejorar o reducir la susceptibilidad a una condición clínica como se describe en este documento. En una realización preferida, el término tratamiento se refiere a un tratamiento profiláctico (es decir, una terapia para reducir la susceptibilidad a una afección clínica) , de un trastorno o afección como se define en el presente documento. Por tanto, "tratamiento", "tratar" y sus términos equivalentes se refieren a la obtención de un efecto farmacológico o fisiológico deseado, que cubre cualquier tratamiento de una afección o trastorno patológico en un mamífero, incluido un ser humano. El efecto puede ser profiláctico en términos de prevención total o parcial de un trastorno o síntoma del mismo y / o puede ser terapéutico en términos de una cura parcial o completa de un trastorno y / o efecto adverso atribuible al trastorno. Es decir, "tratamiento" incluye (1) evitar que el trastorno ocurra o reaparezca en un sujeto, (2) inhibir el trastorno, como detener su desarrollo, (3) detener o terminar el trastorno o, al menos, los síntomas asociados con el mismo, de modo que el huésped ya no sufra el trastorno o sus síntomas, como provocar la regresión del trastorno o sus síntomas, por ejemplo, al restaurar o reparar una función perdida, altante o defectuosa, o estimular un proceso ineficiente, o (4) aliviar, aliviar o mejorar el trastorno o los síntomas asociados con el mismo. El término "sujeto" como se usa en el presente documento, se refiere a cualquier sujeto, particularmente un sujeto mamífero, para el que se desea la terapia. Los sujetos mamíferos incluyen humanos, animales domésticos, animales de granja y zoológicos, deportes o animales de compañía como perros, gatos, cobayas, conejos, ratas, ratones, caballos, ganado, vacas, etc. En una realización preferida de la invención, el sujeto es un mamífero. En una realización más preferida de la invención, el sujeto es un ser humano. La administración de los compuestos, composiciones o formas farmacéuticas de la presente invención puede ser realizada mediante cualquier método adecuado, como la infusión intravenosa y las vías oral, tópica o parenteral. La administración oral es la preferida por la conveniencia de los pacientes. La cantidad administrada de un compuesto de la presente invención dependerá de la relativa eficacia del compuesto elegido, la severidad de la enfermedad a tratar y el peso del paciente. Sin embargo, los compuestos de esta invención serán administrados una o más veces al día, por ejemplo 1, 2, 3 ó 4 veces diarias, con una dosis total entre 0.1 y 1000 mg/Kg/día. Es importante tener en cuenta que puede ser necesario introducir variaciones en la dosis, dependiendo de la edad y de la condición del paciente, así como modificaciones en la vía de administración. A lo largo de la descripción y las reivindicaciones la palabra "comprende" y sus variantes no pretenden excluir otras características técnicas, aditivos, componentes o pasos. Para los expertos en la materia, otros objetos, ventajas y características de la invención se desprenderán en parte de la descripción y en parte de la práctica de la invención. Los siguientes ejemplos y figuras se proporcionan a modo de ilustración, y no se pretende que sean limitativos de la presente invención. EJEMPLOS DE LA INVENCIÓN Cribado virtual Con el objetivo de encontrar nuevos inhibidores para la hepsina se realizó un cribado virtual mediante la técnica de docking molecular a partir de la estructura cristalográfica de la proteína epositada en la Protein Data Bank con código 1P57 frente a la librería de compuestos DrugBank (https://go.drugbank.com/) en las coordenadas de su sitio catalítico, caracterizada por residuos HIS57, ASP102 y SER195. El docking molecular se realizó en un clúster de cónputo de alto rendimiento mediante el software Autodock Vina 1.1.2 (http://vina.scripps.edu/) y para ello se convirtieron tanto la estructura de la proteína como la librería de ligandos DrugBank al formato pdbqt mediante MGLTools. Una vez terminados los cálculos se priorizaron los compuestos en función de docking score y se examinaron visualmente los 8 primeros, seleccionando el compuesto Suramin (código DrugBank DB04786 y primero en la lista) con un docking score de -12 Kcal/mol e interaciones hidrofóbicas con los residuos ALA-39, LEU-41, HIS-57, PRO-60, PHE-97, GLN-151, interacciónes mediante puente de hidrogeno con los residuos GLY-38, HIS-40, HIS-57, ASN-63, GLN-73, ASN-99, GLN-151, GLY-153, SER-195, SER-214, GLY-216, GLY-219, interacciones por "puentes de sal" con ARG-34, HIS-57 y una interacción "piStacking" con el residuo HIS-57 como se aprecia en la Figura 1. En dicha figura podemos observar que Suramin impide el acceso a la triada catalítica, representada en formato de superficie para sus tres residuos Suramin actúa como inhibidor irreversible de hepsina. Mediante un ensayo fluorogénico se determinó la capacidad de Suramin de inhibir la actividad proteolítica de hepsina. Hepsina es una serín proteasas capaz de hidrolizar al sustrato BOC-Gln-Arg-Arg-AMC (Bachem, Barcelona, Spain) , de manera que es posible registrar la emisión de fluorescencia mediante un lector de placas con longitudes de onda de excitación y emisión de 380 nm y 460 nm, respectivamente. Para ello, se incubó hepsina (0.05 j M) (R&D Systems, Madrid, Spain) en tampón 50 mM Tris-HCl, pH 9 buffer, con 200 j M BOC-Gln-Arg-Arg-AMC y se registró la fluorescencia emitida durante 5 minutos. Para comprobar el efecto de Suramin, se llevó a cabo la misma reacción, pero incubando previamente hepsina con Suramin durante 1 hora a 37°C y en distintas concentraciones que oscilaron entre 0.13 y 10 |jM. De esta forma se calculó el valor de IC50, o concentración de inhibidor a la que se alcanza la mitad de la velocidad máxima de hepsina en la hidrólisis de su sustrato, que fue de 0.66 j M. Suramin reduce la migración de células de cáncer colorrectal Una vez que las células alcanzan confluencia, forman una monocapa. Mediante la eliminación de una tira de células 300-500 ^m de anchura con una punta de pipetea de 200 ^l, es posible evaluar la capacidad de migración de las células y el efecto de Suramín en este proceso. La capacidad de migración se evalúa en función del porcentaje de área ocupada tras 48 horas de incubación y se cuantifica utilizando el software ImageJ. Los resultados muestran que Suramín a una concentración de 0, 66 ^M reduce de forma significativa la migración de células Caco-2, tanto con expresión basal como con sobreexpresión de hepsina, mediante transfección estable de un plásmido que contiene el gen HPN. Suramin reduce la invasión de células de cáncer colorrectal Para evaluar la invasión se realizó un ensayo en el que se evaluó la capacidad de las células para degradar una matriz de gelatina. Para ello, se mezcló gelatina al 0.2% y rodamina (Invitrogen, Life Technologies, Madrid, Spain) , en una ratio 1:55 en tampón NaCl 61 mM, borohidrato sódico 50 mM y después se dializó toda la noche en PBS. Se prepararon cubres con la mezcla preparada cubriéndolos y se fijó la matriz con glutaraldehido 0.5% durante 15 min. Los cubres se lavaron con PBS y se añadieron las suspensiones de células Caco-2 durante 72 horas. Posteriormente se fijaron las células con formaldehído 3.7% y se incubó con phalloidin faloidin?? (0, 01 mg/ml) y ProLongGold Antifade con DAPI (4', 6-diamino-2-phenylindole; ThermoFisher) . Las imágenes se tomaron con un microscopio confocal SP8 LEICA y se analizaron con ImageJ. Los resultados mostraron que Suramín, a una concentración de 0, 66 ^M, reduce de forma significativa la degradación de la gelatina de células Caco-2 tanto con expresión basal como con sobreexpresión de hepsina. Suramin reduce la generación de trombina Puesto que hepsina activa al FVII y, por tanto, la cascada de la coagulación, podría contribuir al estado de hipercoagulabilidad de los pacientes con cáncer colorrectal con niveles elevados de hepsina. Para comprobar la hipótesis, se incubó plasma procedente de 20 sujetos sanos, a los que se extrajo sangre citratada 3.8% mediante venopunción, con las células Caco-2 con sobreexpresión de hepsina. El plasma se centrifugó previamente a 2500 g 20 minutos. Para el ensayo de generación de trombina, se retiró el plasma incubado y se incubó con el reactivo LOW® (factor tisular: 1 pmol; fosfolípidos: 4 ^mol; Diagnostica Stago) en una placa de 96 pocillo. Todas las muestras se prepararon en duplicado. La coagulación en estas muestras se inició añadiendo cloruro cálcico en un tampón que contenía el sustrato fluorogénico FluCa-kit reagent® (Diagnostica Stago) . Para cada muestra individual se utilizó un calibrador de rombina. La fluorescencia se registró durante 60 min en un fluorímetro Fluoroskan Ascent (Thermolab Systems) y los datos se analizaron utilizando el software Thrombinoscope™ (version 5.0.0.742; Diagnostica Stago) . Se analizaron los siguientes parámetros: (a) lag-time, que indica el inicio de la fase de generación de trombina; (b) tiempo hasta alcanzar la máxima concentración de trombina (ttPeak) ; (c) concentración máxima de trombina (Peak) ; (d) índice de frecuencia media (MRI) de la fase de propagación de la generación de trombina calculada por la fórmula Peak/ (ttPeak - lag-time) y se expresa en nM/min; y (e) potencial endógeno de trombina (ETP) que muestra la actividad enzimática de trombina evaluada como el área bajo la curva. Los resultados muestran que Suramín reduce el potencial endógeno de trombina (ETP) , el pico máximo (peak) y la velocidad en alcanzar el pico mientras que provoca un incremento en el tiempo de inicio de la fase de generación de trombina (lag-time) y el tiempo en alcanzar el pico. Tabla 1. Valores promedios del test de generación de trombina. Suramin no afecta ni a la muerte celular, ni a la proliferación celular Se quiso evaluar si la menor migración o invasión celular podría deberse a una mayor muerte, o a una menor proliferación celular. Para ello se incubaron las células (5x105 cells/ml) en placas de 6 pocillos en presencia o ausencia de Suramín tras 48 h. El tipo de muerte celular se determinó mediante el ensayo de anexina V/7-ADD (Anexin V Apoptosis Detection Kit FITC; eBiosciences, Thermo Fisher, Karlsruhe, Alemania; 7-ADD BD-Biosciences) siguiendo las instrucciones del fabricante. Para evaluar la actividad proliferativa de las células Caco-2, se añadió 5-etinil-2'-desoxiuridina (EdU) (Thermo Fisher Scientific) durante 48 h. Luego, se usaron alícuotas de las células tumorales para determinar el porcentaje de células positivas, es decir, Caco-2 o Caco-2-HPN-EdU+, detectadas por reacción de acoplamiento de azida luorescente con EdU de acuerdo con el protocolo del fabricante (Click-iT; Thermo Fisher Scientific) . Los resultados mostraron que Suramín no afecta ni a la muerte (porcentajes entorno al 5-10 %) ni a la proliferación celular. Suramin inhibe la invasión de las células de cáncer colorrectal en modelos de xenotrasplante de larvas de pez cebra Se obtuvieron peces cebra wild type (Danio rerio H. Cypriniformes, Cyprinidae) del Zebrafish International Resource Center (ZIRC, Oregon, EE. UU.) Los peces roya9/a9; nacrew2/w2 (casper) transparentes se obtuvieron como ya se ha descrito previamente en la bibliografía [R.M. White, A. Sessa, C. Burke, T. Bowman, J. LeBlanc, C. Ceol, et al. Transparent adult zebrafish as a tool for in vivo transplantation analysis. Cell Stem Cell 2008;2:183-9]. Los peces se aparearon, clasificaron, criaron y procesaron como se describe en el manual del pez cebra [M. Westerfield. The zebrafish book: a guide for the laborator y use of zebrafish. Http//Zfin Org/Zf_info/Zfbook/Zfbk Html 2000.]. Los huevos de pez cebra fertilizados se obtuvieron del desove natural y se mantuvieron en nuestras instalaciones siguiendo las prácticas estándar de cría. Los animales se mantuvieron en un ciclo de luz / oscuridad de 12 h a 28 ° C. Las larvas de pez cebra se anestesiaron con 0.16 mg/ml ethyl 3-aminobenzoato (tricaine; Sigma-Aldrich) . Las células Caco-2 y Caco-2-HPN se cultivaron en presencia y ausencia de 0, 66 ^M de suramin durante 48 h, luego se disgregaron y marcaron con 1, 1'-di-octa-decil-3, 3, 3 ', 3'-perclorato de tetra-metil-indo-carbo-cianina (Dil, ThermoFisher) y finalmente se resuspendieron en un tampón que contenía suero bovino fetal al 5% en PBS. La inyección de las células en embriones se realizó mediante la inyección de 200 células/embrión en el saco vitelino de larvas de pez cebra wild type o Casper 48 horas después de la fertilización y después de 5 días a 35°C, las larvas se analizaron mediante microscopía de fluorescencia para comprobar la diseminación de las células tumorales. La puntuación de la invasión de células Caco-2 y Caco-2-HPN se calculó como el porcentaje de larvas con invasión de células tumorales con respecto al total de larvas analizadas. El pretratamiento de las células Caco-2-HPN (CACO HPN) con suramin (HPN + S) redujo su capacidad de invasión a los niveles encontrados en las células parentales (CACO WT) (Figura 8) , lo que confirma los estudios in vitro y muestra además su potencial terapéutico para el tratamiento del cáncer colorrectal.
Publicaciones:
ES2912350 (25/05/2022) - A1 Solicitud de patente con informe sobre el estado de la técnica
ES2912350 (13/11/2023) - B2 Patente de invención con examen
ES2912350 (05/12/2023) - B9 Patente de invención corregida
Eventos:
En fecha 19/09/2021 se realizó Registro Instancia de Solicitud
En fecha 19/09/2021 se realizó Admisión a Trámite
En fecha 19/09/2021 se realizó Aceptación Tramitación CAP
En fecha 19/09/2021 se realizó 1001P_Comunicación Admisión a Trámite
En fecha 22/09/2021 se realizó Superado examen de oficio
En fecha 13/05/2022 se realizó Realizado IET
En fecha 17/05/2022 se realizó 1109P_Comunicación Traslado del IET
En fecha 25/05/2022 se realizó Publicación Solicitud
En fecha 25/05/2022 se realizó Publicación Folleto Solicitud con IET (A1)
En fecha 24/08/2022 se realizó PETEX_Petición de examen sustantivo
En fecha 09/09/2022 se realizó Validación petición y/o pago de examen sustantivo conforme
En fecha 29/11/2022 se realizó El solicitante ha contestado pero existen nuevas objeciones a la concesión de la solicitud
En fecha 29/11/2022 se realizó Elaboración de examen sustantivo
En fecha 30/11/2022 se realizó 6120P_Notificación de examen sustantivo
En fecha 05/12/2022 se realizó Publicación de examen sustantivo
En fecha 23/01/2023 se realizó No Inscripcion de Cesion F202230730
En fecha 06/02/2023 se realizó 5127P_Subsanación a defectos en examen sustantivo
En fecha 30/03/2023 se realizó 6120P_Notificación de examen sustantivo
En fecha 05/04/2023 se realizó Publicación de examen sustantivo
En fecha 05/06/2023 se realizó 3585X_Registro Solicitud Prórroga de Plazos
En fecha 05/06/2023 se realizó Concesión Prórroga de Plazos
En fecha 05/06/2023 se realizó 1585X_Notificación Concesión Prórroga de Plazos
En fecha 09/06/2023 se realizó Publicación Concesión Prórroga de Plazos (BOPI)
En fecha 07/08/2023 se realizó 5127P_Subsanación a defectos en examen sustantivo
En fecha 19/10/2023 se realizó Designación de Comisión de Expertos
En fecha 20/10/2023 se realizó Finalización de Examen Sustantivo
En fecha 20/10/2023 se realizó 6121P_Comunicación finalización de examen sustantivo
En fecha 26/10/2023 se realizó Publicación finalización de examen sustantivo
En fecha 03/11/2023 se realizó Concesión con examen sustantivo
En fecha 03/11/2023 se realizó Entrega título
En fecha 03/11/2023 se realizó 6125P_Notificación de concesión con examen sustantivo
En fecha 13/11/2023 se realizó Publicación concesión Patente
En fecha 13/11/2023 se realizó Publicación Folleto Concesión
En fecha 05/12/2023 se realizó Publicación de la Patente de invención corregida (BOPI)
En fecha 05/12/2023 se realizó Publicación Folleto Patente deInvención Corregida (B9)
Pagos:
19/09/2021 - Pago Tasas IET
14/12/2023 - Pago 03 Anualidad
+ ES-2912350_B91.- Un compuesto de fórmula (I) o una sal, estereoisómero, tautómero, hidrato o solvato farmacéuticamente aceptable del mismo, para su uso en el tratamiento del cáncer colorrectal caracterizado por que el individuo presenta los niveles de hepsina elevados con respecto a los niveles normales y el cáncer colorrectal es no metastásico. 2.- Una composición que comprende el compuesto de fórmula (I) según la reivindicación anterior, para su uso en el tratamiento del cáncer colorrectal caracterizado por que el individuo presenta los niveles de hepsina elevados con respecto a los niveles normales y el cáncer colorrectal es no metastásico. 3.- La composición para su uso según la reivindicación anterior, que es una composición farmacéutica. 4.- La composición para su uso según cualquiera de las reivindicaciones 2-3, donde la composición además comprende excipientes y/o un vehículo farmacéuticamente aceptable. 5.- La composición para su uso según cualquiera de las reivindicaciones 3-4, que además comprende otro principio activo. 6.- Un kit de partes que comprende: a) Una parte A, que comprende un compuesto o una composición según cualquiera de las reivindicaciones 1-5, y b) Una parte B, que es un agente antineoplásico para su uso en el tratamiento del cáncer colorrectal donde el individuo presenta los niveles de hepsina elevados con respecto a los niveles normales y el cáncer colorrectal es no metastásico 7.- Un kit de partes que comprende: a) Una parte A, que comprende un compuesto o una composición según cualquiera de las reivindicaciones 1-5, y b) Una parte B, que es un agente antineoplásico para su uso en la prevención o el tratamiento de los eventos tromboembólicos venosos desarrollados como consecuencia de la terapia antineoplásica según la reivindicación 6.
+ ES-2912350_B21 Un compuesto de fórmula (I) o una sal, estereoisómero, tautómero, hidrato o solvato farmacéuticamente aceptable del mismo, para su uso en el tratamiento del cáncer colorrectal caracterizado por que el individuo presenta los niveles de hepsina elevados con respecto a los niveles normales y el cáncer colorrectal es no metastásico. 2.- Una composición que comprende el compuesto de fórmula (I) según la reivindicación anterior, para su uso en el tratamiento del cáncer colorrectal caracterizado por que el individuo presenta los niveles de hepsina elevados con respecto a los niveles normales y el cáncer colorrectal es no metastásico. 3.- La composición para su uso según la reivindicación anterior, que es una composición farmacéutica. 4.- La composición para su uso según cualquiera de las reivindicacione s 2-3, donde la composición además comprende excipientes y/o un vehículo farmacéuticamente aceptable. 5.- La composición para su uso según cualquiera de las reivindicaciones 3-4, que además comprende otro principio activo. 6.- Un kit de partes que comprende: a) Una parte A, que comprende un compuesto o una composición según cualquiera de las reivindicaciones 1-5, y b) Una parte B, que es un agente antineoplásico para su uso en el tratamiento del cáncer colorrectal donde el individuo presenta los niveles de hepsina elevados con respecto a los niveles normales y el cáncer colorrectal es no metastásico 7.- Un kit de partes que comprende: a) Una parte A, que comprende un compuesto o una composición según cualquiera de las reivindicaciones 1-5, y b) Una parte B, que es un agente antineoplásico para su uso en la prevención o el tratamiento de los eventos tromboembólicos venosos desarrollados como consecuencia de la terapia antineoplásica según la reivindicación 6.
+ ES-2912350_A11.- Un compuesto de fórmula (I) Fórmula (I) o una sal, estereoisómero, tautómero, hidrato o solvato farmacéuticamente aceptable del mismo, para la prevención y tratamiento del cáncer colorrectal. 2.- El compuesto para su uso según la reivindicación 1, donde el cáncer colorrectal es el cáncer colorrectal metastásico. 3.- El compuesto para su uso según cualquiera de las reivindicaciones 1-2, donde el individuo presenta los niveles de hepsina elevados con respecto a los niveles normales. 4.- Una composición que comprende un compuesto de fórmula (I) según la reivindicación anterior, para su uso según cualquiera de las reivindicaciones 1-3. 5.- La composición según la reivindicación anterior, que es una composición farmacéutica. 6.- La composición según cualquiera de las reivindicaciones 4-5, donde la composición además comprende excipientes y/o un vehículo farmacéuticamente aceptable. 7.- La composición según cualquiera de las reivindicaciones 4-6, que además comprende otro principio activo. 8.- Un kit de partes que comprende: a) Una parte A, que comprende un compuesto o una composición según cualquiera de las reivindicaciones 1-7, y b) Una parte B, que es un agente antineoplásico. 9.- El kit de partes según la reivindicación 8 para la prevención o el tratamiento de los eventos tromboembólicos venosos (ETEV) desarrollados como consecuencia de la terapia antineoplásica.
Los productos y servicios protegidos por este registro son:
A61K 31/185 - A61P 35/00 - A61P 35/04 - A61P 7/02
Descripciones:
+ ES-2912350_B9 Nuevo tratamiento del cáncer colorrectal CAMPO DE LA INVENCIÓN La presente invención pertenece al campo de la biomedicina, y más concretamente se refiere a un nuevo tratamiento para el cáncer colorrectal. ANTECEDENTES DE LA INVENCIÓN El cáncer colorrectal es el tercer cáncer más común diagnosticado tanto en hombres como en mujeres. Tiene lugar en el colon y recto y se desarrolla lentamente a partir de pólipos adenomatosos. El crecimiento excesivo de la mucosa del colon genera estos pólipos que tienen una morfología pedunculada o sésil. El desarrollo de cáncer colorrectal puede tener lugar por diferentes vías. Entre ellas se encuentra la aparición de lesiones serradas con apariencia del epitelio en dientes de sierra y que ocurre en el colon proximal. Esta vía se detecta en un tercio de todos los cánceres colorrectales. Con respecto a la secuencia adenoma-carcinoma, se ha propuesto que la progresión desde adenomas pequeños a grades adenomas o adenocarcinomas avanzados está mediada por múltiples rutas moleculares que incluyen la inestabilidad de microsatélites, la inestabilidad cromosómica y rutas epigenéticas (Jebelli, Asiyeh et al. Medicinal research reviews vol. 41, 1 (2021) : 395-434) . Es posible hacer una prevención precoz puesto que se ha estimado que al menos lleva 10 años que un pólipo se transforme en célula tumoral (0ines, Mari et al. "Epidemiology and risk factors of colorectal polyps." Best practice & research. Clinical gastroenterology vol. 31, 4 (2017) : 419-424) . El diagnóstico precoz mediante colonoscopia y detección de sangre oculta en heces, así como la mejora en los tratamientos ha contribuido a prolongar la supervivencia de los tumores colorrectales en la fase de curación, Sin embargo, la metástasis está presente en aproximadamente el 25% de los pacientes durante el diagnóstico y en total, el 50% de los pacientes con cáncer colorrectal desarrollará metástasis (Ohhara, Yoshihito et al. World journal of gastrointestinal oncology vol. 8, 9 (2016) : 642-55) . Debido a la heterogeneidad y naturaleza multifactorial del cáncer colorrectal han aumentado considerablemente en los últimos años estudios para identificar predictores clínicos y moleculares así como factores pronósticos con potencial para mejorar el manejo de los pacientes (Aran, Veronica et al. Clinical colorectal cancer vol. 15, 3 (2016) : 195-203) . Tanto la incidencia como la mortalidad del cáncer colorrectal ha disminuido tanto para hombres como para mujeres desde 1970 debido a la detección precoz. Sin embargo, aunque diferentes terapias estás disponibles para el cáncer colorrectal metastásico, los resultados no son óptimos debido a la heterogeneidad entre pacientes debido a características moleculares, respuesta a la terapia y presentación clínica (Linnekamp, Janneke F et al. Cancer research vol. 75, 2 (2015) : 245-9) . Los tratamientos más comunes de estos tumores se basan en la cirugía, la quimioterapia, terapias dirigidas y el tratamiento hormonal sustitutivo (Rejhová, A et al. European journal of medicinal chemistr y vol. 144 (2018) : 582-594) . Al menos el 50% de los pacientes con cáncer colorrectal recaen tras la resección quirúrgica y finalmente mueren de enfermedad metastásica. A pesar de la quimioterapia adyuvante postoperatoria en estos pacientes para disminuir el riesgo de recurrencia, el tratamiento adyuvante no da beneficios en supervivencia, especialmente en pacientes diagnosticados con estadío I (Aran, Veronica et al. Clinical colorectal cancer vol. 15, 3 (2016) : 195-203) . La terapia dirigida está basada en anticuerpos monoclonales y pequeños inhibidores moleculares, que selectivamente bloquean la proliferación celular mediante la intervención con ciertas moléculas y proteínas sobreexpresadas requeridas para la expansión del tumor (Jebelli, Asiyeh et al. Medicinal research reviews vol. 41, 1 (2021) : 395-434) . Las proteasas pericelulares han sido ampliamente implicadas en carcinogénesis, no solo porque actúan como enzimas capaces de degradar la matriz extracelular, permitiendo así a las células tumorales romper la membrana basal e invadir el tejido circundante, sino que son capaces de actuar como modificadores proteolíticos de factores de crecimiento y receptores activados por proteasas que son críticos para la activación de rutas de señalización tumoral (Tanabe, Lauren M, and Karin List. The FEBS journal vol. 284, 10 (2017) : 1421-1436) . Entre estas proteasas se encuentra hepsina, que pertenece a la super familia de serín proteasas transmembrana de tipo II (TTSP) . La Hepsina se encuentra sobreexpresada en cáncer de próstata y sus niveles elevados son indicativos de mal pronóstico y recaída tras prostatectomía radical (Sardana, Girish et al. Clinical chemistr y vol. 54, 12 (2008) : 1951-60.) . También se ha demostrado su sobreexpresión en cáncer de ovario (Miao, Jiangyong et al. International journal of cancer vol. 123, 9 (2008) : 2041-7) y cáncer de mama (Miao, Jiangyong et al. International journal of cancer vol. 123, 9 (2008) : 2041-7) . El desarrollo de eventos tromboembólicos venosos (ETEV) es un problema común en los tumores digestivos. La incidencia acumulada de ETEV en series modernas varía entre 12 20% (Posch, Florian et al. Thrombosis and haemostasis vol. 115, 4 (2016) : 817-26) , dependiendo de las disparidades en las características de los pacientes en cada estudio y el contexto en el que la evaluación es llevada a cabo. La trombogénesis se atribuye a una combinación de factores clínicos y biológicos, que incluyen la activación del sistema hemostático por parte del tumor y la toxicidad vascular de la quimioterapia. La trombosis se asocia con una mayor morbilidad y mortalidad en estos pacientes (Carmona-Bayonas, A et al. British journal of cancer vol. 116, 8 (2017) : 994-1001) y es uno de los eventos adversos más importantes en los ensayos clínicos de terapia anti-neoplásica. BREVE DESCRIPCIÓN DE LOS DIBUJOS Fig. 1. Fórmula de Suramina. Fig. 2. Pose resultante del docking molecular entre Suramina y hepsina. Fig. 3. Cálculo del valor de IC50 de Suramina sobre la actividad de Hepsina. Velocidad máxima de la actividad de Hepsina en presencia de dosis crecientes de Suramina. Fig. 4. Efecto de Suramina en la migración de células de cáncer colorrectal. Porcentaje de migración celular de las células Caco-2 y Caco-2-HPN en presencia o ausencia de Suramina. Fig. 5. Efecto de Suramina en la degración de gelatina de células de cáncer colorrectal. Porcentaje de degradación celular de las células Caco-2 y Caco-2-HPN en presencia o ausencia de Suramina. Fig. 6. Efecto de Suramina en la generación de trombina. Valores del potencial endógeno de trombina en células Caco-2 y Caco-2-HPN. Valores del tiempo hasta el inicio del pico (Lagtime) , yel tiempo en alcanzar el pico máximo (ttPeak) en las células Caco-2-HPN en presencia o ausencia de Suramina. Fig. 7. Efecto de Suramina en la proliferación de células de cáncer colorrectal. Porcentaje de células Caco-2 y Caco-2-HPN que están proliferando (EdU+) en presencia o ausencia de Suramina. Fig. 8. Efecto del pretratamiento con Suramina de células Caco-2-HPN (HPN-S) en la capacidad de invasión. DESCRIPCIÓN DE LA INVENCIÓN Los autores de la presente invención han demostrado que el Suramina reduce la migración de células de cáncer colorrectal, reduce la invasión de células de cáncer colorrectal, reduce la generación de trombina en plasma procedente de sujetos sanos tras la incubación con células de cáncer colorrectal y reduce la generación de trombina en plasma de pacientes con cáncer colorrectal con elevados niveles de hepsina en plasma. Han demostrado que el suramina, un fármaco utilizado para el tratamiento de la tripanosomiasis africana, la oncocercosis y en investigación para el tratamiento del cáncer de próstata (PMID: 15484217) , es inhibidor irreversible de hepsina. Por tanto, un primer aspecto de la invención se refiere al compuesto de fórmula (I) Fórmula (I) o cualquiera de sus sales, preferiblemente cualquier sal farmacéuticamente aceptable, ésteres, tautómeros, polimorfos, hidratos farmacéuticamente aceptables, o un isómero, profármacos, derivados, solvatos o análogos, o cualquiera de sus combinaciones, para su uso en el tratamiento del cáncer colorrectal caracterizado por que el individuo presenta los niveles de hepsina elevados con respecto a los niveles normales, y el cáncer colorrectal es cáncer no metastásico Como se puede observar en la Fig. 4, el Suramina es capaz de disminuir la migración de las células del cáncer colorrectal. Además, reduce de forma significativa la degradación de la gelatina de células Caco-2 tanto con expresión basal como con sobreexpresión de hepsina (Fig. 5) . Tal y como se muestra en la Fig. 6, el compuesto de la invención, reduce la generación de trombina en plasma de pacientes con cáncer colorrectal con elevados niveles de hepsina en plasma. En una realización particular, el compuesto de la invención se emplea para la prevención o tratamiento de los eventos tromboembólicos venosos (ETEV) , y preferiblemente de aquellos desarrollados como consecuencia de la terapia antineoplásica. Los compuestos de fórmula (I) , o sus sales o solvatos para uso de acuerdo con la invención están preferiblemente en forma farmacéuticamente aceptable o sustancialmente pura. Por forma farmacéuticamente aceptable se entiende, entre otras cosas, que tiene un nivel de pureza farmacéuticamente aceptable excluyendo los aditivos farmacéuticos normales tales como diluyentes y vehículos, y que no incluye ningún material considerado tóxico a niveles de dosificación normales. Los niveles de pureza de la sustancia farmacéutica están preferiblemente por encima del 50%, más preferiblemente por encima del 70%, lo más preferiblemente por encima del 90%. En una realización preferida, está por encima del 95% del compuesto de fórmula (I) o de su sal, estereoisómero o solvato farmacéuticamente aceptable. La invención también se refiere a usos según la invención de metabolitos de los compuestos descritos en la presente descripción. Un "metabolito" de un compuesto descrito en el presente documento es un derivado de ese compuesto que se forma cuando el compuesto se metaboliza. El término "metabolito activo" se refiere a un derivado biológicamente activo de un compuesto que se forma cuando el compuesto se metaboliza. El término "metabolizado", como se usa en este documento, se refiere a la suma de los procesos (que incluyen, pero no se limitan a, reacciones de hidrólisis y reacciones catalizadas por enzimas) mediante los cuales un organismo cambia una sustancia particular. Por tanto, las enzimas pueden producir alteraciones estructurales específicas en un compuesto. Después de entrar en el cuerpo, la mayoría de los medicamentos son sustratos para reacciones químicas que pueden cambiar us propiedades físicas y efectos biológicos. Estas conversiones metabólicas, que suelen afectar a la polaridad de los compuestos de la invención, alteran la forma en que los fármacos se distribuyen y excretan en el organismo. Sin embargo, en algunos casos, se requiere el metabolismo de un fármaco para obtener un efecto terapéutico. La invención también se refiere a profármacos de los compuestos para uso según la invención. El término "prodroga" o "profármaco" tal como aquí se utiliza incluye cualquier derivado de un compuesto de fórmula (I) , -por ejemplo y no limitativamente: ésteres (incluyendo ésteres de ácidos carboxílicos, ésteres de aminoácidos, ésteres de fosfato, ésteres de sulfonato de sales metálicas, etc.) , carbamatos, amidas, etc.- que al ser administrado a un individuo puede ser transformado directa o indirectamente en dicho compuesto de fórmula (I) en el mencionado individuo. Ventajosamente, dicho derivado es un compuesto que aumenta la biodisponibilidad del compuesto de fórmula (I) cuando se administra a un individuo o que potencia la liberación del compuesto de fórmula (I) en un compartimento biológico. La naturaleza de dicho derivado no es crítica siempre y cuando pueda ser administrado a un individuo y proporcione el compuesto de fórmula (I) , en un compartimento biológico de un individuo. La preparación de dicho profármaco puede llevarse a cabo mediante métodos convencionales conocidos por los expertos en la materia, que son capaces de liberar el compuesto de fórmula (I) como ingrediente activo cuando el profármaco se administra a un sujeto mamífero. La liberación del ingrediente activo ocurre in vivo. Los profármacos se pueden preparar mediante técnicas conocidas por los expertos en la técnica. Estas técnicas generalmente modifican los grupos funcionales apropiados en un compuesto dado. Sin embargo, estos grupos funcionales modificados regeneran grupos funcionales originales mediante manipulación rutinaria o in vivo. Los profármacos de los compuestos de la invención incluyen compuestos en los que se modifica un grupo hidroxi, amino, carboxílico o similar. Los ejemplos de profármacos incluyen, pero no se limitan a, ésteres. Los compuestos de fórmula general (I) y sus profármacos que contienen uno o varios centros quirales pueden estar presentes como racematos, mezclas diastereoméricas o isómeros individuales ópticamente activos. Los compuestos de la presente invención representados por la fórmula (I) pueden incluir isómeros, dependiendo de la presencia de enlaces múltiples, incluyendo isómeros ópticos o enantiómeros, dependiendo de la presencia de centros quirales. Los isómeros, enantiómeros o diastereoisómeros individuales y las mezclas de los mismos caen dentro del alcance de la presente invención, es decir, el término isómero también se refiere a cualquier mezcla de isómeros, como diastereómeros, racémicos, etc., incluso a sus isómeros ópticamente activos o las mezclas en distintas proporciones de los mismos. Los nantiómeros o diastereoisómeros individuales, así como sus mezclas, pueden separarse mediante técnicas convencionales. Composiciones y composiciones farmacéuticas El compuesto de fórmula (I) de la invención puede estar formando parte de una composición, preferiblemente de una composición farmacéutica. Otro aspecto de la invención se refiere a una composición que comprende el compuesto de fórmula (I) de la invención, de ahora en adelante composición de la invención, o una sal, estereoisómero, tautómero, hidrato o solvato farmacéuticamente aceptable del mismo, para la prevención y tratamiento del cáncer colorrectal caracterizado por que el individuo presenta los niveles de hepsina elevados con respecto a los niveles normales, y el cáncer colorrectal es cáncer no metastásico. En otra realización particular, se emplea el compuesto de la invención para la prevención o tratamiento de los eventos tromboembólicos venosos (ETEV) , y preferiblemente de aquellos desarrollados como consecuencia de la terapia antineoplásica. En otra realización de este aspecto, la composición de la invención además comprende otro principio activo. Más preferiblemente es una composición farmacéutica. En otra realización preferida además comprende vehículos farmacéuticamente aceptables. En otra realización preferida de este aspecto, la primera composición de la invención solo comprende como principio activo el compuesto de fórmula (I) , aunque puede comprender, además, excipientes y vehículos farmacéuticamente aceptables. El término "medicamento", tal y como se usa en esta memoria, hace referencia a cualquier sustancia usada para prevención, diagnóstico, alivio, tratamiento o curación de enfermedades en el hombre y los animales. La administración de los compuestos, composiciones o formas farmacéuticas de la presente invención puede ser realizada mediante cualquier método adecuado, como la infusión intravenosa y las vías oral, tópica o parenteral. La administración oral es la preferida por la conveniencia de los pacientes. La cantidad administrada de un compuesto de la presente invención dependerá de la relativa eficacia del compuesto elegido, la severidad de la enfermedad a tratar y el peso del paciente. Sin embargo, los compuestos de esta invención serán administrados una o más veces al día, por ejemplo 1, 2, 3 O 4 veces diarias, con una dosis total entre 0.1 y 1000 mg/Kg/día. Es importante tener en cuenta que puede ser necesario introducir variaciones en la dosis, dependiendo de la edad y de la condición del paciente, así como modificaciones en la vía de administración. "Composición farmacéutica", como se usa en el presente documento, se refiere a composiciones y entidades moleculares que son fisiológicamente tolerables y no producen típicamente una reacción alérgica o una reacción desfavorable similar a como trastornos gástricos, mareos y similares, cuando se administra a un humano o animal. Preferiblemente, el término "farmacéuticamente aceptable" significa que está aprobado por una agencia reguladora de un gobierno estatal o federal o está incluido en la Farmacopea de los Estados Unidos u otra farmacopea generalmente reconocida para uso en animales, y más particularmente en humanos. El término "excipiente" se refiere a un vehículo, diluyente o adyuvante que se administra con el ingrediente activo. Dichos excipientes farmacéuticos pueden ser líquidos estériles, como agua y aceites, incluidos los de origen petrolífero, animal, vegetal o sintético, como aceite de cacahuete, aceite de soja, aceite mineral, aceite de sésamo y similares. Como vehículos se utilizan preferiblemente agua o soluciones acuosas salinas y soluciones acuosas de dextrosa y glicerol, particularmente para soluciones inyectables. Los vehículos farmacéuticos adecuados se describen en "Remington's Phamaceutical Sciences" de EW Martin, 21a edición, 2005; o "Manual de excipientes farmacéuticos", Rowe CR; Paul JS; Marian EQ, sexta edición. Como se emplea aquí, el término "principio activo", "substancia activa", "substancia farmacéuticamente activa", "ingrediente activo" ó "ingrediente farmacéuticamente activo" significa cualquier componente que potencialmente proporcione una actividad farmacológica u otro efecto diferente en el diagnóstico, cura, mitigación, tratamiento, o prevención de una enfermedad, o que afecta a la estructura o función del cuerpo del hombre u otros animales. El término incluye aquellos componentes que promueven un cambio químico en la elaboración del fármaco y están presentes en el mismo de una forma modificada prevista que proporciona la actividad específica o el efecto. Las cantidades apropiadas de un compuesto de fórmula (I) como se define anteriormente, o una sal, estereoisómero farmacéuticamente aceptable o solvato del mismo se pueden formular con excipientes y/o vehículos farmacéuticamente aceptables para obtener una composición farmacéutica para uso en los usos médicos de la invención. Los vehículos farmacéuticamente aceptables adecuados incluyen, por ejemplo, agua, soluciones salinas, alcohol, aceites vegetales, polietilenglicoles, gelatina, lactosa, amilosa, estearato de magnesio, talco, tensioactivos, ácido silícico, parafina viscosa, aceite perfumado, monoglicéridos y diglicéridos. de ácidos grasos, ésteres de ácidos grasos, petroetrales, hidroximetilcelulosa, polivinilpirrolidona y similares. Las composiciones farmacéuticas que contienen el compuesto de fórmula (I) , como se definió anteriormente, o una sal, estereoisómero o solvato farmacéuticamente aceptable del mismo para su uso de acuerdo con la invención pueden ocurrir en cualquier forma farmacéutica de administración considerada apropiada para la vía de administración seleccionada, por ejemplo, por administración sistémica (por ejemplo, inyección intravenosa, subcutánea, intramuscular) , oral, parenteral o tópica, para lo cual incluirá los excipientes farmacéuticamente aceptables necesarios para la formulación del método de administración deseado. Además, también es posible administrar la composición que comprende el compuesto de fórmula (I) como se definió anteriormente, o una sal farmacéuticamente aceptable, estereoisómero o solvato del mismo para uso según la invención por vía intranasal o sublingual que permite la administración sistémica mediante un modo de administración no agresivo. Además, la administración intraventricular puede ser adecuada. Una vía de administración preferida es la oral. Cuando sea necesario, el compuesto de fórmula (I) como se definió anteriormente, o una sal, estereoisómero o solvato farmacéuticamente aceptable del mismo para su uso de acuerdo con la invención está comprendido en una composición que también incluye un agente solubilizante y un anestésico local para aliviar cualquier dolor en el lugar de la inyección. Generalmente, los ingredientes se suministran por separado o mezclados en forma de dosificación unitaria, por ejemplo, como un polvo liofilizado seco o un concentrado libre de agua en un recipiente herméticamente cerrado como una ampolla o bolsita que indica la cantidad de agente activo. Cuando la composición deba administrarse mediante infusión, se puede dispensar con una botella de infusión que contenga agua o solución salina estériles de calidad farmacéutica. En casos distintos de la administración intravenosa, la composición puede contener cantidades menores de agentes humectantes o emulsionantes, o agentes tamponantes del pH. La composición puede ser una solución líquida, suspensión, emulsión, gel, polímero o formulación de liberación sostenida. La composición se puede formular con aglutinantes y vehículos tradicionales, como se conoce en la técnica. Las formulaciones pueden incluir vehículos estándar tales como grados farmacéuticos de manitol, lactosa, almidón, estearato de magnesio, sacárido de sodio, celulosa, carbonato de magnesio, etc., vehículos inertes que tienen una funcionalidad bien establecida en la fabricación de productos farmacéuticos. Se conocen varios sistemas de administración y se pueden usar para administrar un compuesto de la presente invención que incluye la encapsulación en liposomas, micropartículas, microcápsulas y similares. Otro aspecto de la invención se refiere a una forma farmacéutica, de ahora en adelante forma farmacéutica de la invención, que comprende un compuesto de la invención (un compuesto de fórmula (I) , o la composición de la invención. En esta memoria se entiende por "forma farmacéutica" la mezcla de uno o más principios activos con o sin aditivos que presentan características físicas para su adecuada dosificación, conservación, administración y biodisponibilidad. Las formas de dosificación sólidas para administración oral pueden incluir cápsulas convencionales, cápsulas de liberación sostenida, comprimidos convencionales, comprimidos, comprimidos masticables, comprimidos sublinguales, efervescentes comprimidos, píldoras, suspensiones, polvos, gránulos y geles de liberación sostenida. En estas formas de dosificación sólidas, los compuestos activos se pueden mezclar con al menos un excipiente inerte como sacarosa, lactosa o almidón. Tales formas de dosificación también pueden comprender, como en la práctica normal, sustancias adicionales distintas de diluyentes inertes, por ejemplo, agentes lubricantes tales como estearato de magnesio. En el caso de cápsulas, comprimidos, comprimidos efervescentes y píldoras, las formas de dosificación también pueden comprender agentes tamponantes. Los comprimidos y las píldoras se pueden preparar con recubrimientos entéricos. Las formas de dosificación líquidas para administración oral pueden incluir emulsiones, soluciones, suspensiones, jarabes y elixires farmacéuticamente aceptables que contienen diluyentes inertes usados comúnmente en la técnica, tales como agua. Esas composiciones ambién pueden comprender adyuvantes tales como agentes humectantes, agentes emulsionantes y de suspensión y agentes edulcorantes, agentes aromatizantes y perfumantes. Las preparaciones inyectables, por ejemplo, suspensiones acuosas u oleaginosas, inyectables estériles se pueden formular de acuerdo con la técnica conocida usando agentes dispersantes, agentes humectantes y/o agentes de suspensión adecuados. Entre los vehículos y disolventes aceptables que se pueden utilizar se encuentran el agua, la solución de Ringer y la solución isotónica de cloruro de sodio. Los aceites estériles también se utilizan convencionalmente como disolventes o medios de suspensión. Para la administración tópica, los compuestos de la invención pueden formularse como cremas, geles, lociones, líquidos, pomadas, rocíe soluciones, dispersiones, barras sólidas, emulsiones, microemulsiones y similares que se pueden formular de acuerdo con métodos convencionales que utilizan excipientes adecuados, tales como, por ejemplo, emulsionantes, tensioactivos, agentes espesantes, agentes colorantes y combinaciones de dos o más de los mismos. Adicionalmente, el compuesto de fórmula (I) como se definió anteriormente, o una sal farmacéuticamente aceptable, estereoisómero o solvato del mismo para uso según la invención se pueden administrar en forma de parches transdérmicos o iontoforesis dispositivos. En una realización, los compuestos para uso según la invención se administran como un parche transdérmico, por ejemplo, en forma de parche transdérmico de liberación sostenida. Se conocen en la técnica parches transdérmicos adecuados. Varios sistemas de suministro de fármacos son conocidos y se pueden utilizar para administrar los agentes o composiciones para uso de acuerdo con la invención, incluyendo, por ejemplo, encapsulación en liposomas, microburbujas, emulsiones, micropartículas, microcápsulas y similares. La dosis requerida se puede administrar como una sola unidad o en una forma de liberación sostenida. Las formas de liberación sostenible y los materiales y métodos apropiados para su preparación se describen, por ejemplo, en " Modified-Release Drug Deliver y Technology ", Rathbone, MJ Hadgraft, J. y Roberts, MS (eds.) , Marcel Dekker, Inc., Nueva York (2002) , " Handbook of Pharmaceutical Controlled Release Technology ", Wise, DL (ed.) , Marcel Dekker, Inc. Nueva York, (2000) . En una realización de la invención, la forma administrable por vía ral de un compuesto para uso según la invención está en una forma de liberación sostenida que comprende además al menos un recubrimiento o matriz. El recubrimiento o matriz de liberación sostenida incluye, sin limitación, polímeros naturales, ceras, grasas, semisintéticos o sintéticos insolubles en agua, modificados, alcoholes grasos, ácidos grasos, plastificantes naturales semisintéticos o sintéticos, o una combinación de dos o más de ellos. Los recubrimientos entéricos se pueden aplicar usando procesos convencionales conocidos por los expertos en la técnica, como se describe en, por ejemplo, Johnson, JL, " Pharmaceutical tablet coating", Coatings Technology Handbook (Segunda edición) , Satas, D. y Tracton, AA (eds. ) , Marcel Dekker, Inc. Nueva York, (2001) , Carstensen, T., " Coating Tablets in Advanced Pharmaceutical Solids", Swarbrick, J. (ed.) , Marcel Dekker, Inc. Nueva York (2001) , 455-468. El término "prevención", "prevenir" o "prevenir", como se usa en el presente documento, se refiere a la administración de un compuesto de acuerdo con la invención o de un medicamento que comprende dicho compuesto a un sujeto que no ha sido diagnosticado como posiblemente tener una enfermedad, pero que normalmente se esperaría que la desarrolle o que tenga un mayor riesgo de padecerla. La prevención pretende evitar la aparición de dicha enfermedad. La prevención puede ser completa (por ejemplo, la ausencia total de una enfermedad) . La prevención también puede ser parcial, de modo que, por ejemplo, la aparición de una enfermedad en un sujeto es menor que la que habría ocurrido sin la administración del compuesto de la presente invención. La prevención también se refiere a la reducción de la susceptibilidad a una condición clínica. El término "tratamiento", como se usa en este documento, se refiere a cualquier tipo de terapia, que tiene como objetivo terminar, prevenir, mejorar o reducir la susceptibilidad a una condición clínica como se describe en este documento. En una realización preferida, el término tratamiento se refiere a un tratamiento profiláctico (es decir, una terapia para reducir la susceptibilidad a una afección clínica) , de un trastorno o afección como se define en el presente documento. Por tanto, "tratamiento", "tratar" y sus términos equivalentes se refieren a la obtención de un efecto farmacológico o fisiológico deseado, que cubre cualquier tratamiento de una afección o trastorno patológico en un mamífero, incluido un ser humano. El efecto puede ser profiláctico en términos de prevención total o parcial de un trastorno o síntoma del mismo y / o puede ser terapéutico en términos de una cura parcial o completa de un trastorno y / o efecto adverso atribuible al trastorno. Es decir, "tratamiento" incluye (1) evitar que el trastorno ocurra o reaparezca en un sujeto, (2) inhibir el trastorno, como detener su desarrollo, (3) detener o terminar el trastorno o, al menos, los síntomas asociados con el mismo, de modo que el huésped ya no sufra el trastorno o sus síntomas, como provocar la regresión del trastorno o sus síntomas, por ejemplo, al restaurar o reparar una función perdida, altante o defectuosa, o estimular un proceso ineficiente, o (4) aliviar, aliviar o mejorar el trastorno o los síntomas asociados con el mismo. El término "sujeto" como se usa en el presente documento, se refiere a cualquier sujeto, particularmente un sujeto mamífero, para el que se desea la terapia. Los sujetos mamíferos incluyen humanos, animales domésticos, animales de granja y zoológicos, deportes o animales de compañía como perros, gatos, cobayas, conejos, ratas, ratones, caballos, ganado, vacas, etc. En una realización preferida de la invención, el sujeto es un mamífero. En una realización más preferida de la invención, el sujeto es un ser humano. La administración de los compuestos, composiciones o formas farmacéuticas de la presente invención puede ser realizada mediante cualquier método adecuado, como la infusión intravenosa y las vías oral, tópica o parenteral. La administración oral es la preferida por la conveniencia de los pacientes. La cantidad administrada de un compuesto de la presente invención dependerá de la relativa eficacia del compuesto elegido, la severidad de la enfermedad a tratar y el peso del paciente. Sin embargo, los compuestos de esta invención serán administrados una o más veces al día, por ejemplo 1, 2, 3 ó 4 veces diarias, con una dosis total entre 0.1 y 1000 mg/Kg/día. Es importante tener en cuenta que puede ser necesario introducir variaciones en la dosis, dependiendo de la edad y de la condición del paciente, así como modificaciones en la vía de administración. A lo largo de la descripción y las reivindicaciones la palabra "comprende" y sus variantes no pretenden excluir otras características técnicas, aditivos, componentes o pasos. Para los expertos en la materia, otros objetos, ventajas y características de la invención se desprenderán en parte de la descripción y en parte de la práctica de la invención. Los siguientes ejemplos y figuras se proporcionan a modo de ilustración, y no se pretende que sean limitativos de la presente invención. EJEMPLOS DE LA INVENCIÓN Cribado virtual Con el objetivo de encontrar nuevos inhibidores para la hepsina se realizó un cribado virtual mediante la técnica de docking molecular a partir de la estructura cristalográfica de la proteína epositada en la Protein Data Bank con código 1P57 frente a la librería de compuestos DrugBank (https://go.drugbank.com/) en las coordenadas de su sitio catalítico, caracterizada por residuos HIS57, ASP102 y SER195. El docking molecular se realizó en un clúster de cómputo de alto rendimiento mediante el software Autodock Vina 1.1.2 ( http://vina.scripps.edu/) y para ello se convirtieron tanto la estructura de la proteína como la librería de ligandos DrugBank al formato pdbqt mediante MGLTools. Una vez terminados los cálculos se priorizaron los compuestos en función de docking score y se examinaron visualmente los 8 primeros, seleccionando el compuesto Suramina (código DrugBank DB04786 y primero en la lista) con un docking score de -12 Kcal/mol e interaciones hidrofóbicas con los residuos ALA-39, LEU-41, HIS-57, PRO-60, PHE-97, GLN-151, interacciónes mediante puente de hidrogeno con los residuos GLY-38, HIS-40, HIS-57, ASN-63, GLN-73, ASN-99, GLN-151, GLY-153, SER-195, SER-214, GLY-216, GLY-219, interacciones por "puentes de sal" con ARG-34, HIS-57 y una interacción "piStacking" con el residuo HIS-57 como se aprecia en la Figura 1. En dicha figura podemos observar que Suramina impide el acceso a la triada catalítica, representada en formato de superficie para sus tres residuos Suramina actúa como inhibidor irreversible de hepsina. Mediante un ensayo fluorogénico se determinó la capacidad de Suramina de inhibir la actividad proteolítica de hepsina. Hepsina es una serín proteasas capaz de hidrolizar al sustrato BOC-Gln-Arg-Arg-AMC (Bachem, Barcelona, Spain) , de manera que es posible registrar la emisión de fluorescencia mediante un lector de placas con longitudes de onda de excitación y emisión de 380 nm y 460 nm, respectivamente. Para ello, se incubó hepsina (0.05 j M) (R&D Systems, Madrid, Spain) en tampón 50 mM Tris-HCl, pH 9 buffer, con 200 |jM BOC-Gln-Arg-Arg-AMC y se registró la fluorescencia emitida durante 5 minutos. Para comprobar el efecto de Suramina, se llevó a cabo la misma reacción, pero incubando previamente hepsina con Suramina durante 1 hora a 37°C y en distintas concentraciones que oscilaron entre 0.13 y 10 j M. De esta forma se calculó el valor de IC50, o concentración de inhibidor a la que se alcanza la mitad de la velocidad máxima de hepsina en la hidrólisis de su sustrato, que fue de 0.66 j M. Suramina reduce la migración de células de cáncer colorrectal Una vez que las células alcanzan confluencia, forman una monocapa. Mediante la eliminación de una tira de células 300-500 ^m de anchura con una punta de pipetea de 200 ^l, es posible evaluar la capacidad de migración de las células y el efecto de Suramina en este proceso. La capacidad de migración se evalúa en función del porcentaje de área ocupada tras 48 horas de incubación y se cuantifica utilizando el software ImageJ. Los resultados muestran que Suramina a una concentración de 0, 66 ^M reduce de forma significativa la migración de células Caco-2, tanto con expresión basal como con sobreexpresión de hepsina, mediante transfección estable de un plásmido que contiene el gen HPN. Suramina reduce la invasión de células de cáncer colorrectal Para evaluar la invasión se realizó un ensayo en el que se evaluó la capacidad de las células para degradar una matriz de gelatina. Para ello, se mezcló gelatina al 0.2% y rodamina (Invitrogen, Life Technologies, Madrid, Spain) , en una ratio 1:55 en tampón NaCl 61 mM, borohidrato sódico 50 mM y después se dializó toda la noche en PBS. Se prepararon cubres con la mezcla preparada cubriéndolos y se fijó la matriz con glutaraldehido 0.5% durante 15 min. Los cubres se lavaron con PBS y se añadieron las suspensiones de células Caco-2 durante 72 horas. Posteriormente se fijaron las células con formaldehído 3.7% y se incubó con phalloidin faloidin?? (0, 01 mg/ml) y ProLongGold Antifade con DAPI (4', 6-diamino-2-phenylindole; ThermoFisher) . Las imágenes se tomaron con un microscopio confocal SP8 LEICA y se analizaron con ImageJ. Los resultados mostraron que Suramina, a una concentración de 0, 66 ^M, reduce de forma significativa la degradación de la gelatina de células Caco-2 tanto con expresión basal como con sobreexpresión de hepsina. Suramina reduce la generación de trombina Puesto que hepsina activa al FVII y, por tanto, la cascada de la coagulación, podría contribuir al estado de hipercoagulabilidad de los pacientes con cáncer colorrectal con niveles elevados de hepsina. Para comprobar la hipótesis, se incubó plasma procedente de 20 sujetos sanos, a los que se extrajo sangre citratada 3.8% mediante venopunción, con las células Caco-2 con sobreexpresión de hepsina. El plasma se centrifugó previamente a 2500 g 20 minutos. Para el ensayo de generación de trombina, se retiró el plasma incubado y se incubó con el reactivo LOW® (factor tisular: 1 pmol; fosfolípidos: 4 ^mol; Diagnostica Stago) en una placa de 96 pocillo. Todas las muestras se prepararon en duplicado. La coagulación en estas muestras se inició añadiendo cloruro cálcico en un tampón que contenía el sustrato fluorogénico FluCa-kit reagent® (Diagnostica Stago) . Para cada muestra individual se utilizó un calibrador de rombina. La fluorescencia se registró durante 60 min en un fluorímetro Fluoroskan Ascent (Thermolab Systems) y los datos se analizaron utilizando el software Thrombinoscope™ (version 5.0.0.742; Diagnostica Stago) . Se analizaron los siguientes parámetros: (a) lag-time, que indica el inicio de la fase de generación de trombina; (b) tiempo hasta alcanzar la máxima concentración de trombina (ttPeak) ; (c) concentración máxima de trombina (Peak) ; (d) índice de frecuencia media (MRI) de la fase de propagación de la generación de trombina calculada por la fórmula Peak/ (ttPeak - lag-time) y se expresa en nM/min; y (e) potencial endógeno de trombina (ETP) que muestra la actividad enzimática de trombina evaluada como el área bajo la curva. Los resultados muestran que Suramina reduce el potencial endógeno de trombina (ETP) , el pico máximo (peak) y la velocidad en alcanzar el pico mientras que provoca un incremento en el tiempo de inicio de la fase de generación de trombina (lag-time) y el tiempo en alcanzar el pico. Tabla 1. Valores promedios del test de generación de trombina. Suramina no afecta ni a la muerte celular, ni a la proliferación celular Se quiso evaluar si la menor migración o invasión celular podría deberse a una mayor muerte, o a una menor proliferación celular. Para ello se incubaron las células (5x105 cells/ml) en placas de 6 pocillos en presencia o ausencia de Suramina tras 48 h. El tipo de muerte celular se determinó mediante el ensayo de anexina V/7-ADD (Anexin V Apoptosis Detection Kit FITC; eBiosciences, Thermo Fisher, Karlsruhe, Alemania; 7-ADD BD-Biosciences) siguiendo las instrucciones del fabricante. Para evaluar la actividad proliferativa de las células Caco-2, se añadió 5-etinil-2'-desoxiuridina (EdU) (Thermo Fisher Scientific) durante 48 h. Luego, se usaron alícuotas de las células tumorales para determinar el porcentaje de células positivas, es decir, Caco-2 o Caco-2-HPN-EdU+, detectadas por reacción de acoplamiento de azida luorescente con EdU de acuerdo con el protocolo del fabricante (Click-iT; Thermo Fisher Scientific) . Los resultados mostraron que Suramina no afecta ni a la muerte (porcentajes entorno al 5-10 %) ni a la proliferación celular. Suramina inhibe la invasión de las células de cáncer colorrectal en modelos de xenotrasplante de larvas de pez cebra Se obtuvieron peces cebra wild type (Danio rerio H. Cypriniformes, Cyprinidae) del Zebrafish International Resource Center (ZIRC, Oregon, EE. UU.) Los peces roya9/a9; nacrew2/w2 (casper) transparentes se obtuvieron como ya se ha descrito previamente en la bibliografía [R.M. White, A. Sessa, C. Burke, T. Bowman, J. LeBlanc, C. Ceol, et al. Transparent adult zebrafish as a tool for in vivo transplantation analysis. Cell Stem Cell 2008;2:183-9]. Los peces se aparearon, clasificaron, criaron y procesaron como se describe en el manual del pez cebra [M. Westerfield. The zebrafish book: a guide for the laborator y use of zebrafish. Http//Zfin Org/Zf_info/Zfbook/Zfbk Html 2000.]. Los huevos de pez cebra fertilizados se obtuvieron del desove natural y se mantuvieron en nuestras instalaciones siguiendo las prácticas estándar de cría. Los animales se mantuvieron en un ciclo de luz / oscuridad de 12 h a 28 ° C. Las larvas de pez cebra se anestesiaron con 0.16 mg/ml ethyl 3-aminobenzoato (tricaine; Sigma-Aldrich) . Las células Caco-2 y Caco-2-HPN se cultivaron en presencia y ausencia de 0, 66 ^M de suramina durante 48 h, luego se disgregaron y marcaron con 1, 1'-di-octa-decil-3, 3, 3 ', 3'-perclorato de tetra-metil-indo-carbo-cianina (Dil, ThermoFisher) y finalmente se resuspendieron en un tampón que contenía suero bovino fetal al 5% en PBS. La inyección de las células en embriones se realizó mediante la inyección de 200 células/embrión en el saco vitelino de larvas de pez cebra wild type o Casper 48 horas después de la fertilización y después de 5 días a 35°C, las larvas se analizaron mediante microscopía de fluorescencia para comprobar la diseminación de las células tumorales. La puntuación de la invasión de células Caco-2 y Caco-2-HPN se calculó como el porcentaje de larvas con invasión de células tumorales con respecto al total de larvas analizadas. El pretratamiento de las células Caco-2-HPN (CACO HPN) con suramina (HPN + S) redujo su capacidad de invasión a los niveles encontrados en las células parentales (CACO WT) (Figura 8) , lo que confirma los estudios in vitro y muestra además su potencial terapéutico para el tratamiento del cáncer colorrectal.
+ ES-2912350_B2 Nuevo tratamiento del cáncer colorrectal CAMPO DE LA INVENCIÓN La presente invención pertenece al campo de la biomedicina, y más concretamente se refiere a un nuevo tratamiento para el cáncer colorrectal. ANTECEDENTES DE LA INVENCIÓN El cáncer colorrectal es el tercer cáncer más común diagnosticado tanto en hombres como en mujeres. Tiene lugar en el colon y recto y se desarrolla lentamente a partir de pólipos adenomatosos. El crecimiento excesivo de la mucosa del colon genera estos pólipos que tienen una morfología pedunculada o sésil. El desarrollo de cáncer colorrectal puede tener lugar por diferentes vías. Entre ellas se encuentra la aparición de lesiones serradas con apariencia del epitelio en dientes de sierra y que ocurre en el colon proximal. Esta vía se detecta en un tercio de todos los cánceres colorrectales. Con respecto a la secuencia adenoma-carcinoma, se ha propuesto que la progresión desde adenomas pequeños a grades adenomas o adenocarcinomas avanzados está mediada por múltiples rutas moleculares que incluyen la inestabilidad de microsatélites, la inestabilidad cromosómica y rutas epigenéticas (Jebelli, Asiyeh et al. Medicinal research reviews vol. 41, 1 (2021) : 395-434) . Es posible hacer una prevención precoz puesto que se ha estimado que al menos lleva 10 años que un pólipo se transforme en célula tumoral (0ines, Mari et al. "Epidemiology and risk factors of colorectal polyps." Best practice & research. Clinical gastroenterology vol. 31, 4 (2017) : 419-424) . El diagnóstico precoz mediante colonoscopia y detección de sangre oculta en heces, así como la mejora en los tratamientos ha contribuido a prolongar la supervivencia de los tumores colorrectales en la fase de curación, Sin embargo, la metástasis está presente en aproximadamente el 25% de los pacientes durante el diagnóstico y en total, el 50% de los pacientes con cáncer colorrectal desarrollará metástasis (Ohhara, Yoshihito et al. World journal of gastrointestinal oncology vol. 8, 9 (2016) : 642-55) . Debido a la heterogeneidad y naturaleza multifactorial del cáncer colorrectal han aumentado considerablemente en los últimos años estudios para identificar predictores clínicos y moleculares así como factores pronósticos con potencial para mejorar el manejo de los pacientes (Aran, Veronica et al. Clinical colorectal cancer vol. 15, 3 (2016) : 195-203) . Tanto la incidencia como la mortalidad del cáncer colorrectal ha disminuido tanto para hombres como para mujeres desde 1970 debido a la detección precoz. Sin embargo, aunque diferentes terapias estás disponibles para el cáncer colorrectal metastásico, los resultados no son óptimos debido a la heterogeneidad entre pacientes debido a características moleculares, respuesta a la terapia y presentación clínica (Linnekamp, Janneke F et al. Cancer research vol. 75, 2 (2015) : 245-9) . Los tratamientos más comunes de estos tumores se basan en la cirugía, la quimioterapia, terapias dirigidas y el tratamiento hormonal sustitutivo (Rejhová, A et al. European journal of medicinal chemistr y vol. 144 (2018) : 582-594) . Al menos el 50% de los pacientes con cáncer colorrectal recaen tras la resección quirúrgica y finalmente mueren de enfermedad metastásica. A pesar de la quimioterapia adyuvante postoperatoria en estos pacientes para disminuir el riesgo de recurrencia, el tratamiento adyuvante no da beneficios en supervivencia, especialmente en pacientes diagnosticados con estadío I (Aran, Veronica et al. Clinical colorectal cancer vol. 15, 3 (2016) : 195-203) . La terapia dirigida está basada en anticuerpos monoclonales y pequeños inhibidores moleculares, que selectivamente bloquean la proliferación celular mediante la intervención con ciertas moléculas y proteínas sobreexpresadas requeridas para la expansión del tumor (Jebelli, Asiyeh et al. Medicinal research reviews vol. 41, 1 (2021) : 395-434) . Las proteasas pericelulares han sido ampliamente implicadas en carcinogénesis, no solo porque actúan como enzimas capaces de degradar la matriz extracelular, permitiendo así a las células tumorales romper la membrana basal e invadir el tejido circundante, sino que son capaces de actuar como modificadores proteolíticos de factores de crecimiento y receptores activados por proteasas que son críticos para la activación de rutas de señalización tumoral (Tanabe, Lauren M, and Karin List. The FEBS journal vol. 284, 10 (2017) : 1421-1436) . Entre estas proteasas se encuentra hepsina, que pertenece a la super familia de serín proteasas transmembrana de tipo II (TTSP) . La Hepsina se encuentra sobreexpresada en cáncer de próstata y sus niveles elevados son indicativos de mal pronóstico y recaída tras prostatectomía radical (Sardana, Girish et al. Clinical chemistr y vol. 54, 12 (2008) : 1951-60.) . También se ha demostrado su sobreexpresión en cáncer de ovario (Miao, Jiangyong et al. International journal of cancer vol. 123, 9 (2008) : 2041-7) y cáncer de mama (Miao, Jiangyong et al. International journal of cancer vol. 123, 9 (2008) : 2041-7) . El desarrollo de eventos tromboembólicos venosos (ETEV) es un problema común en los tumores digestivos. La incidencia acumulada de ETEV en series modernas varía entre 12 20% (Posch, Florian et al. Thrombosis and haemostasis vol. 115, 4 (2016) : 817-26) , dependiendo de las disparidades en las características de los pacientes en cada estudio y el contexto en el que la evaluación es llevada a cabo. La trombogénesis se atribuye a una combinación de factores clínicos y biológicos, que incluyen la activación del sistema hemostático por parte del tumor y la toxicidad vascular de la quimioterapia. La trombosis se asocia con una mayor morbilidad y mortalidad en estos pacientes (Carmona-Bayonas, A et al. British journal of cancer vol. 116, 8 (2017) : 994-1001) y es uno de los eventos adversos más importantes en los ensayos clínicos de terapia anti-neoplásica. BREVE DESCRIPCIÓN DE LOS DIBUJOS Fig. 1. Fórmula de Suramina. Fig. 2. Pose resultante del docking molecular entre Suramina y hepsina. Fig. 3. Cálculo del valor de IC50 de Suramina sobre la actividad de Hepsina. Velocidad máxima de la actividad de Hepsina en presencia de dosis crecientes de Suramina. Fig. 4. Efecto de Suramina en la migración de células de cáncer colorrectal. Porcentaje de migración celular de las células Caco-2 y Caco-2-HPN en presencia o ausencia de Suramina. Fig. 5. Efecto de Suramina en la degración de gelatina de células de cáncer colorrectal. Porcentaje de degradación celular de las células Caco-2 y Caco-2-HPN en presencia o ausencia de Suramina. Fig. 6. Efecto de Suramina en la generación de trombina. Valores del potencial endógeno de trombina en células Caco-2 y Caco-2-HPN. Valores del tiempo hasta el inicio del pico (Lagtime) , yel tiempo en alcanzar el pico máximo (ttPeak) en las células Caco-2-HPN en presencia o ausencia de Suramina. Fig. 7. Efecto de Suramina en la proliferación de células de cáncer colorrectal. Porcentaje de células Caco-2 y Caco-2-HPN que están proliferando (EdU+) en presencia o ausencia de Suramina. Fig. 8. Efecto del pretratamiento con Suramina de células Caco-2-HPN (HPN-S) en la capacidad de invasión. DESCRIPCIÓN DE LA INVENCIÓN Los autores de la presente invención han demostrado que el Suramina reduce la migración de células de cáncer colorrectal, reduce la invasión de células de cáncer colorrectal, reduce la generación de trombina en plasma procedente de sujetos sanos tras la incubación con células de cáncer colorrectal y reduce la generación de trombina en plasma de pacientes con cáncer colorrectal con elevados niveles de hepsina en plasma. Han demostrado que el suramina, un fármaco utilizado para el tratamiento de la tripanosomiasis africana, la oncocercosis y en investigación para el tratamiento del cáncer de próstata (PMID: 15484217) , es inhibidor irreversible de hepsina. Por tanto, un primer aspecto de la invención se refiere al compuesto de fórmula (I) Fórmula (I) o cualquiera de sus sales, preferiblemente cualquier sal farmacéuticamente aceptable, ésteres, tautómeros, polimorfos, hidratos farmacéuticamente aceptables, o un isómero, profármacos, derivados, solvatos o análogos, o cualquiera de sus combinaciones, para su uso en el tratamiento del cáncer colorrectal caracterizado por que el individuo presenta los niveles de hepsina elevados con respecto a los niveles normales, y el cáncer colorrectal es cáncer no metastásico Como se puede observar en la Fig. 4, el Suramina es capaz de disminuir la migración de las células del cáncer colorrectal. Además, reduce de forma significativa la degradación de la gelatina de células Caco-2 tanto con expresión basal como con sobreexpresión de hepsina (Fig. 5) . Tal y como se muestra en la Fig. 6, el compuesto de la invención, reduce la generación de trombina en plasma de pacientes con cáncer colorrectal con elevados niveles de hepsina en plasma. En una realización particular, el compuesto de la invención se emplea para la prevención o tratamiento de los eventos tromboembólicos venosos (ETEV) , y preferiblemente de aquellos desarrollados como consecuencia de la terapia antineoplásica. Los compuestos de fórmula (I) , o sus sales o solvatos para uso de acuerdo con la invención están preferiblemente en forma farmacéuticamente aceptable o sustancialmente pura. Por forma farmacéuticamente aceptable se entiende, entre otras cosas, que tiene un nivel de pureza farmacéuticamente aceptable excluyendo los aditivos farmacéuticos normales tales como diluyentes y vehículos, y que no incluye ningún material considerado tóxico a niveles de dosificación normales. Los niveles de pureza de la sustancia farmacéutica están preferiblemente por encima del 50%, más preferiblemente por encima del 70%, lo más preferiblemente por encima del 90%. En una realización preferida, está por encima del 95% del compuesto de fórmula (I) o de su sal, estereoisómero o solvato farmacéuticamente aceptable. La invención también se refiere a usos según la invención de metabolitos de los compuestos descritos en la presente descripción. Un "metabolito" de un compuesto descrito en el presente documento es un derivado de ese compuesto que se forma cuando el compuesto se metaboliza. El término "metabolito activo" se refiere a un derivado biológicamente activo de un compuesto que se forma cuando el compuesto se metaboliza. El término "metabolizado", como se usa en este documento, se refiere a la suma de los procesos (que incluyen, pero no se limitan a, reacciones de hidrólisis y reacciones catalizadas por enzimas) mediante los cuales un organismo cambia una sustancia particular. Por tanto, las enzimas pueden producir alteraciones estructurales específicas en un compuesto. Después de entrar en el cuerpo, la mayoría de los medicamentos son sustratos para reacciones químicas que pueden cambiar sus propiedades físicas y efectos biológicos. Estas conversiones metabólicas, que suelen fectar a la polaridad de los compuestos de la invención, alteran la forma en que los fármacos se distribuyen y excretan en el organismo. Sin embargo, en algunos casos, se requiere el metabolismo de un fármaco para obtener un efecto terapéutico. La invención también se refiere a profármacos de los compuestos para uso según la invención. El término "prodroga" o "profármaco" tal como aquí se utiliza incluye cualquier derivado de un compuesto de fórmula (I) , -por ejemplo y no limitativamente: ésteres (incluyendo ésteres de ácidos carboxílicos, ésteres de aminoácidos, ésteres de fosfato, ésteres de sulfonato de sales metálicas, etc.) , carbamatos, amidas, etc.- que al ser administrado a un individuo puede ser transformado directa o indirectamente en dicho compuesto de fórmula (I) en el mencionado individuo. Ventajosamente, dicho derivado es un compuesto que aumenta la biodisponibilidad del compuesto de fórmula (I) cuando se administra a un individuo o que potencia la liberación del compuesto de fórmula (I) en un compartimento biológico. La naturaleza de dicho derivado no es crítica siempre y cuando pueda ser administrado a un individuo y proporcione el compuesto de fórmula (I) , en un compartimento biológico de un individuo. La preparación de dicho profármaco puede llevarse a cabo mediante métodos convencionales conocidos por los expertos en la materia, que son capaces de liberar el compuesto de fórmula (I) como ingrediente activo cuando el profármaco se administra a un sujeto mamífero. La liberación del ingrediente activo ocurre in vivo. Los profármacos se pueden preparar mediante técnicas conocidas por los expertos en la técnica. Estas técnicas generalmente modifican los grupos funcionales apropiados en un compuesto dado. Sin embargo, estos grupos funcionales modificados regeneran grupos funcionales originales mediante manipulación rutinaria o in vivo. Los profármacos de los compuestos de la invención incluyen compuestos en los que se modifica un grupo hidroxi, amino, carboxílico o similar. Los ejemplos de profármacos incluyen, pero no se limitan a, ésteres. Los compuestos de fórmula general (I) y sus profármacos que contienen uno o varios centros quirales pueden estar presentes como racematos, mezclas diastereoméricas o isómeros individuales ópticamente activos. Los compuestos de la presente invención representados por la fórmula (I) pueden incluir isómeros, dependiendo de la presencia de enlaces múltiples, incluyendo isómeros ópticos o enantiómeros, dependiendo de la presencia de centros quirales. Los isómeros, enantiómeros o diastereoisómeros individuales y las mezclas de los mismos caen dentro del alcance de la presente invención, es decir, el término isómero también se refiere a cualquier mezcla de isómeros, como diastereómeros, racémicos, etc., incluso a sus isómeros ópticamente activos o las mezclas en distintas proporciones de los mismos. Los nantiómeros o diastereoisómeros individuales, así como sus mezclas, pueden separarse mediante técnicas convencionales. Composiciones y composiciones farmacéuticas El compuesto de fórmula (I) de la invención puede estar formando parte de una composición, preferiblemente de una composición farmacéutica. Otro aspecto de la invención se refiere a una composición que comprende el compuesto de fórmula (I) de la invención, de ahora en adelante composición de la invención, o una sal, estereoisómero, tautómero, hidrato o solvato farmacéuticamente aceptable del mismo, para la prevención y tratamiento del cáncer colorrectal caracterizado por que el individuo presenta los niveles de hepsina elevados con respecto a los niveles normales, y el cáncer colorrectal es cáncer no metastásico. En otra realización particular, se emplea el compuesto de la invención para la prevención o tratamiento de los eventos tromboembólicos venosos (ETEV) , y preferiblemente de aquellos desarrollados como consecuencia de la terapia antineoplásica. En otra realización de este aspecto, la composición de la invención además comprende otro principio activo. Más preferiblemente es una composición farmacéutica. En otra realización preferida además comprende vehículos farmacéuticamente aceptables. En otra realización preferida de este aspecto, la primera composición de la invención solo comprende como principio activo el compuesto de fórmula (I) , aunque puede comprender, además, excipientes y vehículos farmacéuticamente aceptables. El término "medicamento", tal y como se usa en esta memoria, hace referencia a cualquier sustancia usada para prevención, diagnóstico, alivio, tratamiento o curación de enfermedades en el hombre y los animales. La administración de los compuestos, composiciones o formas farmacéuticas de la presente invención puede ser realizada mediante cualquier método adecuado, como la infusión intravenosa y las vías oral, tópica o parenteral. La administración oral es la preferida por la conveniencia de los pacientes. La cantidad administrada de un compuesto de la presente invención dependerá de la relativa eficacia del compuesto elegido, la severidad de la enfermedad a tratar y el peso del paciente. Sin embargo, los compuestos de esta invención serán administrados una o más veces al día, por ejemplo 1, 2, 3 O 4 veces diarias, con una dosis total entre 0.1 y 1000 mg/Kg/día. Es importante tener en cuenta que puede ser necesario introducir variaciones en la dosis, dependiendo de la edad y de la condición del paciente, así como modificaciones en la vía de administración. "Composición farmacéutica", como se usa en el presente documento, se refiere a composiciones y entidades moleculares que son fisiológicamente tolerables y no producen típicamente una reacción alérgica o una reacción desfavorable similar a como trastornos gástricos, mareos y similares, cuando se administra a un humano o animal. Preferiblemente, el término "farmacéuticamente aceptable" significa que está aprobado por una agencia reguladora de un gobierno estatal o federal o está incluido en la Farmacopea de los Estados Unidos u otra farmacopea generalmente reconocida para uso en animales, y más particularmente en humanos. El término "excipiente" se refiere a un vehículo, diluyente o adyuvante que se administra con el ingrediente activo. Dichos excipientes farmacéuticos pueden ser líquidos estériles, como agua y aceites, incluidos los de origen petrolífero, animal, vegetal o sintético, como aceite de cacahuete, aceite de soja, aceite mineral, aceite de sésamo y similares. Como vehículos se utilizan preferiblemente agua o soluciones acuosas salinas y soluciones acuosas de dextrosa y glicerol, particularmente para soluciones inyectables. Los vehículos farmacéuticos adecuados se describen en "Remington's Pharmaceutical Sciences" de EW Martin, 21a edición, 2005; o "Manual de excipientes farmacéuticos", Rowe CR; Paul JS; Marian EQ, sexta edición. Como se emplea aquí, el término "principio activo", "substancia activa", "substancia farmacéuticamente activa", "ingrediente activo" ó "ingrediente farmacéuticamente activo" significa cualquier componente que potencialmente proporcione una actividad farmacológica u otro efecto diferente en el diagnóstico, cura, mitigación, tratamiento, o prevención de una enfermedad, o que afecta a la estructura o función del cuerpo del hombre u otros animales. El término incluye aquellos componentes que promueven un cambio químico en la elaboración del fármaco y están presentes en el mismo de una forma modificada prevista que proporciona la actividad específica o el efecto. Las cantidades apropiadas de un compuesto de fórmula (I) como se define anteriormente, o una sal, estereoisómero farmacéuticamente aceptable o solvato del mismo se pueden formular con excipientes y/o vehículos farmacéuticamente aceptables para obtener una composición farmacéutica para uso en los usos médicos de la invención. Los vehículos farmacéuticamente aceptables adecuados incluyen, por ejemplo, agua, soluciones salinas, alcohol, aceites vegetales, polietilenglicoles, gelatina, lactosa, amilosa, estearato de magnesio, talco, tensioactivos, ácido silícico, parafina viscosa, aceite perfumado, monoglicéridos y diglicéridos. de ácidos grasos, ésteres de ácidos grasos, petroetrales, hidroximetilcelulosa, polivinilpirrolidona y similares. Las composiciones farmacéuticas que contienen el compuesto de fórmula (I) , como se definió anteriormente, o una sal, estereoisómero o solvato farmacéuticamente aceptable del mismo para su uso de acuerdo con la invención pueden ocurrir en cualquier forma farmacéutica de administración considerada apropiada para la vía de administración seleccionada, por ejemplo, por administración sistémica (por ejemplo, inyección intravenosa, subcutánea, intramuscular) , oral, parenteral o tópica, para lo cual incluirá los excipientes farmacéuticamente aceptables necesarios para la formulación del método de administración deseado. Además, también es posible administrar la composición que comprende el compuesto de fórmula (I) como se definió anteriormente, o una sal farmacéuticamente aceptable, estereoisómero o solvato del mismo para uso según la invención por vía intranasal o sublingual que permite la administración sistémica mediante un modo de administración no agresivo. Además, la administración intraventricular puede ser adecuada. Una vía de administración preferida es la oral. Cuando sea necesario, el compuesto de fórmula (I) como se definió anteriormente, o una sal, estereoisómero o solvato farmacéuticamente aceptable del mismo para su uso de acuerdo con la invención está comprendido en una composición que también incluye un agente solubilizante y un anestésico local para aliviar cualquier dolor en el lugar de la inyección. Generalmente, los ingredientes se suministran por separado o mezclados en forma de dosificación unitaria, por ejemplo, como un polvo liofilizado seco o un concentrado libre de agua en un recipiente herméticamente cerrado como una ampolla o bolsita que indica la cantidad de agente activo. Cuando la composición deba administrarse mediante infusión, se puede dispensar con una botella de infusión que contenga agua o solución salina estériles de calidad farmacéutica. En casos distintos de la administración intravenosa, la composición puede contener cantidades menores de agentes humectantes o emulsionantes, o agentes tamponantes del pH. La composición puede ser una solución líquida, suspensión, emulsión, gel, polímero o formulación de liberación sostenida. La composición se puede formular con aglutinantes y vehículos tradicionales, como se conoce en la técnica. Las formulaciones pueden incluir vehículos estándar tales como grados farmacéuticos de manitol, lactosa, almidón, estearato de magnesio, sacárido de sodio, celulosa, carbonato de magnesio, etc., vehículos inertes que tienen una funcionalidad bien establecida en la fabricación de productos farmacéuticos. Se conocen varios sistemas de administración y se pueden usar para administrar un compuesto de la presente invención que incluye la encapsulación en liposomas, micropartículas, microcápsulas y similares. Otro aspecto de la invención se refiere a una forma farmacéutica, de ahora en adelante forma farmacéutica de la invención, que comprende un compuesto de la invención (un compuesto de fórmula (I) , o la composición de la invención. En esta memoria se entiende por "forma farmacéutica" la mezcla de uno o más principios activos con o sin aditivos que presentan características físicas para su adecuada dosificación, conservación, administración y biodisponibilidad. Las formas de dosificación sólidas para administración oral pueden incluir cápsulas convencionales, cápsulas de liberación sostenida, comprimidos convencionales, comprimidos, comprimidos masticables, comprimidos sublinguales, efervescentes comprimidos, píldoras, suspensiones, polvos, gránulos y geles de liberación sostenida. En estas formas de dosificación sólidas, los compuestos activos se pueden mezclar con al menos un excipiente inerte como sacarosa, lactosa o almidón. Tales formas de dosificación también pueden comprender, como en la práctica normal, sustancias adicionales distintas de diluyentes inertes, por ejemplo, agentes lubricantes tales como estearato de magnesio. En el caso de cápsulas, comprimidos, comprimidos efervescentes y píldoras, las formas de dosificación también pueden comprender agentes tamponantes. Los comprimidos y las píldoras se pueden preparar con recubrimientos entéricos. Las formas de dosificación líquidas para administración oral pueden incluir emulsiones, soluciones, suspensiones, jarabes y elixires farmacéuticamente aceptables que contienen diluyentes inertes usados comúnmente en la técnica, tales como agua. Esas composiciones ambién pueden comprender adyuvantes tales como agentes humectantes, agentes emulsionantes y de suspensión y agentes edulcorantes, agentes aromatizantes y perfumantes. Las preparaciones inyectables, por ejemplo, suspensiones acuosas u oleaginosas, inyectables estériles se pueden formular de acuerdo con la técnica conocida usando agentes dispersantes, agentes humectantes y/o agentes de suspensión adecuados. Entre los vehículos y disolventes aceptables que se pueden utilizar se encuentran el agua, la solución de Ringer y la solución isotónica de cloruro de sodio. Los aceites estériles también se utilizan convencionalmente como disolventes o medios de suspensión. Para la administración tópica, los compuestos de la invención pueden formularse como cremas, geles, lociones, líquidos, pomadas, rocíe soluciones, dispersiones, barras sólidas, emulsiones, microemulsiones y similares que se pueden formular de acuerdo con métodos convencionales que utilizan excipientes adecuados, tales como, por ejemplo, emulsionantes, tensioactivos, agentes espesantes, agentes colorantes y combinaciones de dos o más de los mismos. Adicionalmente, el compuesto de fórmula (I) como se definió anteriormente, o una sal farmacéuticamente aceptable, estereoisómero o solvato del mismo para uso según la invención se pueden administrar en forma de parches transdérmicos o iontoforesis dispositivos. En una realización, los compuestos para uso según la invención se administran como un parche transdérmico, por ejemplo, en forma de parche transdérmico de liberación sostenida. Se conocen en la técnica parches transdérmicos adecuados. Varios sistemas de suministro de fármacos son conocidos y se pueden utilizar para administrar los agentes o composiciones para uso de acuerdo con la invención, incluyendo, por ejemplo, encapsulación en liposomas, microburbujas, emulsiones, micropartículas, microcápsulas y similares. La dosis requerida se puede administrar como una sola unidad o en una forma de liberación sostenida. Las formas de liberación sostenible y los materiales y métodos apropiados para su preparación se describen, por ejemplo, en " Modified-Release Drug Deliver y Technology ", Rathbone, MJ Hadgraft, J. y Roberts, MS (eds.) , Marcel Dekker, Inc., Nueva York (2002) , " Handbook of Pharmaceutical Controlled Release T echnology ", Wise, DL (ed.) , Marcel Dekker, Inc. Nueva York, (2000) . En una realización de la invención, la forma administrable por vía ral de un compuesto para uso según la invención está en una forma de liberación sostenida que comprende además al menos un recubrimiento o matriz. El recubrimiento o matriz de liberación sostenida incluye, sin limitación, polímeros naturales, ceras, grasas, semisintéticos o sintéticos insolubles en agua, modificados, alcoholes grasos, ácidos grasos, plastificantes naturales semisintéticos o sintéticos, o una combinación de dos o más de ellos. Los recubrimientos entéricos se pueden aplicar usando procesos convencionales conocidos por los expertos en la técnica, como se describe en, por ejemplo, Johnson, JL, " Pharmaceutical tablet coating", Coatings Technology Handbook (Segunda edición) , Satas, D. y Tracton, AA (eds. ) , Marcel Dekker, Inc. Nueva York, (2001) , Carstensen, T., " Coating Tablets in Advanced Pharmaceutical Solids", Swarbrick, J. (ed.) , Marcel Dekker, Inc. Nueva York (2001) , 455- 468. El término "prevención", "prevenir" o "prevenir", como se usa en el presente documento, se refiere a la administración de un compuesto de acuerdo con la invención o de un medicamento que comprende dicho compuesto a un sujeto que no ha sido diagnosticado como posiblemente tener una enfermedad, pero que normalmente se esperaría que la desarrolle o que tenga un mayor riesgo de padecerla. La prevención pretende evitar la aparición de dicha enfermedad. La prevención puede ser completa (por ejemplo, la ausencia total de una enfermedad) . La prevención también puede ser parcial, de modo que, por ejemplo, la aparición de una enfermedad en un sujeto es menor que la que habría ocurrido sin la administración del compuesto de la presente invención. La prevención también se refiere a la reducción de la susceptibilidad a una condición clínica. El término "tratamiento", como se usa en este documento, se refiere a cualquier tipo de terapia, que tiene como objetivo terminar, prevenir, mejorar o reducir la susceptibilidad a una condición clínica como se describe en este documento. En una realización preferida, el término tratamiento se refiere a un tratamiento profiláctico (es decir, una terapia para reducir la susceptibilidad a una afección clínica) , de un trastorno o afección como se define en el presente documento. Por tanto, "tratamiento", "tratar" y sus términos equivalentes se refieren a la obtención de un efecto farmacológico o fisiológico deseado, que cubre cualquier tratamiento de una afección o trastorno patológico en un mamífero, incluido un ser humano. El efecto puede ser profiláctico en términos de prevención total o parcial de un trastorno o síntoma del mismo y / o puede ser terapéutico en términos de una cura parcial o completa de un trastorno y / o efecto adverso atribuible al trastorno. Es decir, "tratamiento" incluye (1) evitar que el trastorno ocurra o reaparezca en un sujeto, (2) inhibir el trastorno, como detener su desarrollo, (3) detener o terminar el trastorno o, al menos, los síntomas asociados con el mismo, de modo que el huésped ya no sufra el trastorno o sus síntomas, como provocar la regresión del trastorno o sus síntomas, por ejemplo, al restaurar o reparar una función perdida, altante o defectuosa, o estimular un proceso ineficiente, o (4) aliviar, aliviar o mejorar el trastorno o los síntomas asociados con el mismo. El término "sujeto" como se usa en el presente documento, se refiere a cualquier sujeto, particularmente un sujeto mamífero, para el que se desea la terapia. Los sujetos mamíferos incluyen humanos, animales domésticos, animales de granja y zoológicos, deportes o animales de compañía como perros, gatos, cobayas, conejos, ratas, ratones, caballos, ganado, vacas, etc. En una realización preferida de la invención, el sujeto es un mamífero. En una realización más preferida de la invención, el sujeto es un ser humano. La administración de los compuestos, composiciones o formas farmacéuticas de la presente invención puede ser realizada mediante cualquier método adecuado, como la infusión intravenosa y las vías oral, tópica o parenteral. La administración oral es la preferida por la conveniencia de los pacientes. La cantidad administrada de un compuesto de la presente invención dependerá de la relativa eficacia del compuesto elegido, la severidad de la enfermedad a tratar y el peso del paciente. Sin embargo, los compuestos de esta invención serán administrados una o más veces al día, por ejemplo 1, 2, 3 ó 4 veces diarias, con una dosis total entre 0.1 y 1000 mg/Kg/día. Es importante tener en cuenta que puede ser necesario introducir variaciones en la dosis, dependiendo de la edad y de la condición del paciente, así como modificaciones en la vía de administración. A lo largo de la descripción y las reivindicaciones la palabra "comprende" y sus variantes no pretenden excluir otras características técnicas, aditivos, componentes o pasos. Para los expertos en la materia, otros objetos, ventajas y características de la invención se desprenderán en parte de la descripción y en parte de la práctica de la invención. Los siguientes ejemplos y figuras se proporcionan a modo de ilustración, y no se pretende que sean limitativos de la presente invención. EJEMPLOS DE LA INVENCIÓN Cribado virtual Con el objetivo de encontrar nuevos inhibidores para la hepsina se realizó un cribado virtual mediante la técnica de docking molecular a partir de la estructura cristalográfica de la proteína epositada en la Protein Data Bank con código 1P57 frente a la librería de compuestos DrugBank (https://go.drugbank.com/) en las coordenadas de su sitio catalítico, caracterizada por residuos HIS57, ASP102 y SER195. El docking molecular se realizó en un clúster de cómputo de alto rendimiento mediante el software Autodock Vina 1.1.2 (http://vina.scripps.edu/) y para ello se convirtieron tanto la estructura de la proteína como la librería de ligandos DrugBank al formato pdbqt mediante MGLTools. Una vez terminados los cálculos se priorizaron los compuestos en función de docking score y se examinaron visualmente los 8 primeros, seleccionando el compuesto Suramina (código DrugBank DB04786 y primero en la lista) con un docking score de -12 Kcal/mol e interaciones hidrofóbicas con los residuos ALA-39, LEU-41, HIS-57, PRO-60, PHE-97, GLN-151, interacciónes mediante puente de hidrogeno con los residuos GLY-38, HIS-40, HIS-57, ASN-63, GLN-73, ASN-99, GLN-151, GLY-153, SER-195, SER-214, GLY-216, GLY-219, interacciones por "puentes de sal" con ARG-34, HIS-57 y una interacción "piStacking" con el residuo HIS-57 como se aprecia en la Figura 1. En dicha figura podemos observar que Suramina impide el acceso a la triada catalítica, representada en formato de superficie para sus tres residuos Suramina actúa como inhibidor irreversible de hepsina. Mediante un ensayo fluorogénico se determinó la capacidad de Suramina de inhibir la actividad proteolítica de hepsina. Hepsina es una serín proteasas capaz de hidrolizar al sustrato BOC-Gln-Arg-Arg-AMC (Bachem, Barcelona, Spain) , de manera que es posible registrar la emisión de fluorescencia mediante un lector de placas con longitudes de onda de excitación y emisión de 380 nm y 460 nm, respectivamente. Para ello, se incubó hepsina (0.05 M) (R&D Systems, Madrid, Spain) en tampón 50 mM Tris-HCl, pH 9 buffer, con 200 M BOC-Gln-Arg-Arg-AMC y se registró la fluorescencia emitida durante 5 minutos. Para comprobar el efecto de Suramina, se llevó a cabo la misma reacción, pero incubando previamente hepsina con Suramina durante 1 hora a 37°C y en distintas concentraciones que oscilaron entre 0.13 y 10 M. De esta forma se calculó el valor de IC50, o concentración de inhibidor a la que se alcanza la mitad de la velocidad máxima de hepsina en la hidrólisis de su sustrato, que fue de 0.66 M. Suramina reduce la migración de células de cáncer colorrectal Una vez que las células alcanzan confluencia, forman una monocapa. Mediante la eliminación de una tira de células 300-500 m de anchura con una punta de pipetea de 200 l, es posible evaluar la capacidad de migración de las células y el efecto de Suramina en este proceso. La capacidad de migración se evalúa en función del porcentaje de área ocupada tras 48 horas de incubación y se cuantifica utilizando el software ImageJ. Los resultados muestran que Suramina a una concentración de 0, 66 M reduce de forma significativa la migración de células Caco-2, tanto con expresión basal como con sobreexpresión de hepsina, mediante transfección estable de un plásmido que contiene el gen HPN. Suramina reduce la invasión de células de cáncer colorrectal Para evaluar la invasión se realizó un ensayo en el que se evaluó la capacidad de las células para degradar una matriz de gelatina. Para ello, se mezcló gelatina al 0.2% y rodamina (Invitrogen, Life Technologies, Madrid, Spain) , en una ratio 1:55 en tampón NaCl 61 mM, borohidrato sódico 50 mM y después se dializó toda la noche en PBS. Se prepararon cubres con la mezcla preparada cubriéndolos y se fijó la matriz con glutaraldehido 0.5% durante 15 min. Los cubres se lavaron con PBS y se añadieron las suspensiones de células Caco-2 durante 72 horas. Posteriormente se fijaron las células con formaldehído 3.7% y se incubó con phalloidin faloidin?? (0, 01 mg/ml) y ProLongGold Antifade con DAPI (4', 6-diamino-2-phenylindole; ThermoFisher) . Las imágenes se tomaron con un microscopio confocal SP8 LEICA y se analizaron con ImageJ. Los resultados mostraron que Suramina, a una concentración de 0, 66 M, reduce de forma significativa la degradación de la gelatina de células Caco-2 tanto con expresión basal como con sobreexpresión de hepsina. Suramina reduce la generación de trombina Puesto que hepsina activa al FVII y, por tanto, la cascada de la coagulación, podría contribuir al estado de hipercoagulabilidad de los pacientes con cáncer colorrectal con niveles elevados de hepsina. Para comprobar la hipótesis, se incubó plasma procedente de 20 sujetos sanos, a los que se extrajo sangre citratada 3.8% mediante venopunción, con las células Caco-2 con sobreexpresión de hepsina. El plasma se centrifugó previamente a 2500 g 20 minutos. Para el ensayo de generación de trombina, se retiró el plasma incubado y se incubó con el reactivo LOW® (factor tisular: 1 pmol; fosfolípidos: 4 pmol; Diagnostica Stago) en una placa de 96 pocillo. Todas las muestras se prepararon en duplicado. La coagulación en estas muestras se inició añadiendo cloruro cálcico en un tampón que contenía el sustrato fluorogénico FluCa-kit reagent® (Diagnostica Stago) . Para cada muestra individual se utilizó un calibrador de rombina. La fluorescencia se registró durante 60 min en un fluorímetro Fluoroskan Ascent (Thermolab Systems) y los datos se analizaron utilizando el software Thrombinoscope™ (version 5.0.0.742; Diagnostica Stago) . Se analizaron los siguientes parámetros: (a) lag-time, que indica el inicio de la fase de generación de trombina; (b) tiempo hasta alcanzar la máxima concentración de trombina (ttPeak) ; (c) concentración máxima de trombina (Peak) ; (d) índice de frecuencia media (MRI) de la fase de propagación de la generación de trombina calculada por la fórmula Peak/ (ttPeak - lag-time) y se expresa en nM/min; y (e) potencial endógeno de trombina (ETP) que muestra la actividad enzimática de trombina evaluada como el área bajo la curva. Los resultados muestran que Suramina reduce el potencial endógeno de trombina (ETP) , el pico máximo (peak) y la velocidad en alcanzar el pico mientras que provoca un incremento en el tiempo de inicio de la fase de generación de trombina (lag-time) y el tiempo en alcanzar el pico. Tabla 1. Valores promedios del test de generación de trombina. Suramina no afecta ni a la muerte celular, ni a la proliferación celular Se quiso evaluar si la menor migración o invasión celular podría deberse a una mayor muerte, o a una menor proliferación celular. Para ello se incubaron las células (5x105 cells/ml) en placas de 6 pocillos en presencia o ausencia de Suramina tras 48 h. El tipo de muerte celular se determinó mediante el ensayo de anexina V/7-ADD (Anexin V Apoptosis Detection Kit FITC; eBiosciences, Thermo Fisher, Karlsruhe, Alemania; 7-ADD BD-Biosciences) siguiendo las instrucciones del fabricante. Para evaluar la actividad proliferativa de las células Caco-2, se añadió 5-etinil-2'-desoxiuridina (EdU) (Thermo Fisher Scientific) durante 48 h. Luego, se usaron alícuotas de las células tumorales para determinar el porcentaje de células positivas, es decir, Caco-2 o Caco-2-HPN-EdU+, detectadas por reacción de acoplamiento de azida luorescente con EdU de acuerdo con el protocolo del fabricante (Click-iT; Thermo Fisher Scientific) . Los resultados mostraron que Suramina no afecta ni a la muerte (porcentajes entorno al 5-10 %) ni a la proliferación celular. Suramina inhibe la invasión de las células de cáncer colorrectal en modelos de xenotrasplante de larvas de pez cebra Se obtuvieron peces cebra wild type (Danio rerio H. Cypriniformes, Cyprinidae) del Zebrafish International Resource Center (ZIRC, Oregon, EE. UU.) Los peces roya9/a9; nacrew2/w2 (casper) transparentes se obtuvieron como ya se ha descrito previamente en la bibliografía [R.M. White, A. Sessa, C. Burke, T. Bowman, J. LeBlanc, C. Ceol, et al. Transparent adult zebrafish as a tool for in vivo transplantation analysis. Cell Stem Cell 2008;2:183-9]. Los peces se aparearon, clasificaron, criaron y procesaron como se describe en el manual del pez cebra [M. Westertield. The zebrafish book: a guide for the laborator y use of zebrafish. Http//Zfin Org/Zf_info/Zfbook/Zfbk Html 2000.]. Los huevos de pez cebra fertilizados se obtuvieron del desove natural y se mantuvieron en nuestras instalaciones siguiendo las prácticas estándar de cría. Los animales se mantuvieron en un ciclo de luz / oscuridad de 12 h a 28 ° C. Las larvas de pez cebra se anestesiaron con 0.16 mg/ml ethyl 3-aminobenzoato (tricaine; Sigma-Aldrich) . Las células Caco-2 y Caco-2-HPN se cultivaron en presencia y ausencia de 0, 66 M de suramina durante 48 h, luego se disgregaron y marcaron con 1, 1'-di-octa-decil-3, 3, 3 ', 3'-perclorato de tetra-metil-indo-carbo-cianina (Dil, ThermoFisher) y finalmente se resuspendieron en un tampón que contenía suero bovino fetal al 5% en PBS. La inyección de las células en embriones se realizó mediante la inyección de 200 células/embrión en el saco vitelino de larvas de pez cebra wild type o Casper 48 horas después de la fertilización y después de 5 días a 35°C, las larvas se analizaron mediante microscopía de fluorescencia para comprobar la diseminación de las células tumorales. La puntuación de la invasión de células Caco-2 y Caco-2-HPN se calculó como el porcentaje de larvas con invasión de células tumorales con respecto al total de larvas analizadas. El pretratamiento de las células Caco-2-HPN (CACO HPN) con suramina (HPN + S) redujo su capacidad de invasión a los niveles encontrados en las células parentales (CACO WT) (Figura 8) , lo que confirma los estudios in vitro y muestra además su potencial terapéutico para el tratamiento del cáncer colorrectal.
+ ES-2912350_A1 Nuevo tratamiento del cáncer colorrectal CAMPO DE LA INVENCIÓN La presente invención pertenece al campo de la biomedicina, y más concretamente se refiere a un nuevo tratamiento para el cáncer colorrectal. ANTECEDENTES DE LA INVENCIÓN El cáncer colorrectal es el tercer cáncer más común diagnosticado tanto en hombres como en mujeres. Tiene lugar en el colon y recto y se desarrolla lentamente a partir de pólipos adenomatosos. El crecimiento excesivo de la mucosa del colon genera estos pólipos que tienen una morfología pedunculada o sésil. El desarrollo de cáncer colorrectal puede tener lugar por diferentes vías. Entre ellas se encuentra la aparición de lesiones serradas con apariencia del epitelio en dientes de sierra y que ocurre en el colon proximal. Esta vía se detecta en un tercio de todos los cánceres colorrectales. Con respecto a la secuencia adenoma-carcinoma, se ha propuesto que la progresión desde adenomas pequeños a grades adenomas o adenocarcinomas avanzados está mediada por múltiples rutas moleculares que incluyen la inestabilidad de microsatélites, la inestabilidad cromosómica y rutas epigenéticas (Jebelli, Asiyeh et al. Medicinal research reviews vol. 41, 1 (2021) : 395-434) . Es posible hacer una prevención precoz puesto que se ha estimado que al menos lleva 10 años que un pólipo se transforme en célula tumoral (0ines, Mari et al. "Epidemiology and risk factors of colorectal polyps." Best practice & research. Clinical gastroenterology vol. 31, 4 (2017) : 419-424) . El diagnóstico precoz mediante colonoscopia y detección de sangre oculta en heces, así como la mejora en los tratamientos ha contribuido a prolongar la supervivencia de los tumores colorrectales en la fase de curación, Sin embargo, la metástasis está presente en aproximadamente el 25% de los pacientes durante el diagnóstico y en total, el 50% de los pacientes con cáncer colorrectal desarrollará metástasis (Ohhara, Yoshihito et al. World journal of gastrointestinal oncology vol. 8, 9 (2016) : 642-55) . Debido a la heterogeneidad y naturaleza multifactorial del cáncer colorrectal han aumentado considerablemente en los últimos años estudios para identificar predictores clínicos y moleculares así como factores pronósticos con potencial para mejorar el manejo de los pacientes (Aran, Veronica et al. Clinical colorectal cancer vol. 15, 3 (2016) : 195-203) . Tanto la incidencia como la mortalidad del cáncer colorrectal ha disminuido tanto para hombres como para mujeres desde 1970 debido a la detección precoz. Sin embargo, aunque diferentes terapias estás disponibles para el cáncer colorrectal metastásico, los resultados no son óptimos debido a la heterogeneidad entre pacientes debido a características moleculares, respuesta a la terapia y presentación clínica (Linnekamp, Janneke F et al. Cancer research vol. 75, 2 (2015) : 245-9) . Los tratamientos más comunes de estos tumores se basan en la cirugía, la quimioterapia, terapias dirigidas y el tratamiento hormonal sustitutivo (Rejhová, A et al. European journal of medicinal chemistr y vol. 144 (2018) : 582-594) . Al menos el 50% de los pacientes con cáncer colorrectal recaen tras la resección quirúrgica y finalmente mueren de enfermedad metastásica. A pesar de la quimioterapia adyuvante postoperatoria en estos pacientes para disminuir el riesgo de recurrencia, el tratamiento adyuvante no da beneficios en supervivencia, especialmente en pacientes diagnosticados con estadío I (Aran, Veronica et al. Clinical colorectal cancer vol. 15, 3 (2016) : 195-203) . La terapia dirigida está basada en anticuerpos monoclonales y pequeños inhibidores moleculares, que selectivamente bloquean la proliferación celular mediante la intervención con ciertas moléculas y proteínas sobreexpresadas requeridas para la expansión del tumor (Jebelli, Asiyeh et al. Medicinal research reviews vol. 41, 1 (2021) : 395-434) . Las proteasas pericelulares han sido ampliamente implicadas en carcinogénesis, no solo porque actúan como enzimas capaces de degradar la matriz extracelular, permitiendo así a las células tumorales romper la membrana basal e invadir el tejido circundante, sino que son capaces de actuar como modificadores proteolíticos de factores de crecimiento y receptores activados por proteasas que son críticos para la activación de rutas de señalización tumoral (Tanabe, Lauren M, and Karin List. The FEBS journal vol. 284, 10 (2017) : 1421-1436) . Entre estas proteasas se encuentra hepsina, que pertenece a la super familia de serín proteasas transmembrana de tipo II (TTSP) . La Hepsina se encuentra sobreexpresada en cáncer de próstata y sus niveles elevados son indicativos de mal pronóstico y recaída tras prostatectomía radical (Sardana, Girish et al. Clinical chemistr y vol. 54, 12 (2008) : 1951-60.) . También se ha demostrado su sobreexpresión en cáncer de ovario (Miao, Jiangyong et al. International journal of cancer vol. 123, 9 (2008) : 2041-7) y cáncer de mama (Miao, Jiangyong et al. International journal of cancer vol. 123, 9 (2008) : 2041-7) . El desarrollo de eventos tromboembólicos venosos (ETEV) es un problema común en los tumores digestivos. La incidencia acumulada de ETEV en series modernas varía entre 12 20% (Posch, Florian et al. Thrombosis and haemostasis vol. 115, 4 (2016) : 817-26) , dependiendo de las disparidades en las características de los pacientes en cada estudio y el contexto en el que la evaluación es llevada a cabo. La trombogénesis se atribuye a una combinación de factores clínicos y biológicos, que incluyen la activación del sistema hemostático por parte del tumor y la toxicidad vascular de la quimioterapia. La trombosis se asocia con una mayor morbilidad y mortalidad en estos pacientes (Carmona-Bayonas, A et al. British journal of cancer vol. 116, 8 (2017) : 994-1001) y es uno de los eventos adversos más importantes en los ensayos clínicos de terapia anti-neoplásica. BREVE DESCRIPCIÓN DE LOS DIBUJOS Fig. 1. Fórmula de Suramín. Fig. 2. Pose resultante del docking molecular entre Suramín y hepsina. Fig. 3. Cálculo del valor de IC50 de Suramín sobre la actividad de Hepsina. Velocidad máxima de la actividad de Hepsina en presencia de dosis crecientes de Suramín. Fig. 4. Efecto de Suramín en la migración de células de cáncer colorrectal. Porcentaje de migración celular de las células Caco-2 y Caco-2-HPN en presencia o ausencia de Suramín. Fig. 5. Efecto de Suramín en la degración de gelatina de células de cáncer colorrectal. Porcentaje de degradación celular de las células Caco-2 y Caco-2-HPN en presencia o ausencia de Suramín. Fig. 6. Efecto de Suramín en la generación de trombina. Valores del potencial endógeno de trombina en células Caco-2 y Caco-2-HPN. Valores del tiempo hasta el inicio del pico (Lagtime) , yel tiempo en alcanzar el pico máximo (ttPeak) en las células Caco-2-HPN en presencia o ausencia de Suramín. Fig. 7. Efecto de Suramín en la proliferación de células de cáncer colorrectal. Porcentaje de células Caco-2 y Caco-2-HPN que están proliferando (EdU+) en presencia o ausencia de Suramín. DESCRIPCIÓN DE LA INVENCIÓN Los autores de la presente invención han demostrado que el Suramin reduce la migración de células de cáncer colorrectal, reduce la invasión de células de cáncer colorrectal, reduce la generación de trombina en plasma procedente de sujetos sanos tras la incubación con células de cáncer colorrectal y reduce la generación de trombina en plasma de pacientes con cáncer colorrectal con elevados niveles de hepsina en plasma. Han demostrado que el suramin, un fármaco utilizado para el tratamiento de la tripanosomiasis africana, la oncocercosis y en investigación para el tratamiento del cáncer de próstata (PMID: 15484217) , es inhibidor irreversible de hepsina. Por tanto, un primer aspecto de la invención se refiere al un compuesto de fórmula (I) Fórmula (I) o cualquiera de sus sales, preferiblemente cualquier sal farmacéuticamente aceptable, ésteres, tautómeros, polimorfos, hidratos farmacéuticamente aceptables, o un isómero, profármacos, derivados, solvatos o análogos, o cualquiera de sus combinaciones, para la prevención o tratamiento del cáncer colorrectal. Como se puede observar en la Fig. 4, el Suramin es capaz de disminuir la migración de las células del cáncer colorrectal. Además, reduce de forma significativa la degradación de la gelatina de células Caco-2 tanto con expresión basal como con sobreexpresión de hepsina (Fig. 5) . Por tanto, en una realización preferida de este aspecto de la invención, el cáncer colorrectal es el cáncer colorrectal metastásico. Más preferiblemente, donde el individuo presenta los niveles de hepsina elevados con respecto a los niveles normales. Tal y como se muestra en la Fig. 6, el compuesto de la invención, reduce la generación de trombina en plasma de pacientes con cáncer colorrectal con elevados niveles de hepsina en plasma. Por tanto, otro aspecto de la invención se refiere al compuesto de la invención para la prevención o tratamiento de los eventos tromboembólicos venosos (ETEV) , y preferiblemente de aquellos desarrollados como consecuencia de la terapia antineoplásica. Los compuestos de fórmula (I) , o sus sales o solvatos para uso de acuerdo con la invención están preferiblemente en forma farmacéuticamente aceptable o sustancialmente pura. Por forma farmacéuticamente aceptable se entiende, entre otras cosas, que tiene un nivel de pureza farmacéuticamente aceptable excluyendo los aditivos farmacéuticos normales tales como diluyentes y vehículos, y que no incluye ningún material considerado tóxico a niveles de dosificación normales. Los niveles de pureza de la sustancia farmacéutica están preferiblemente por encima del 50%, más preferiblemente por encima del 70%, lo más preferiblemente por encima del 90%. En una realización preferida, está por encima del 95% del compuesto de fórmula (I) o de su sal, estereoisómero o solvato farmacéuticamente aceptable. La invención también se refiere a usos según la invención de metabolitos de los compuestos descritos en la presente descripción. Un "metabolito" de un compuesto descrito en el presente documento es un derivado de ese compuesto que se forma cuando el compuesto se metaboliza. El término "metabolito activo" se refiere a un derivado biológicamente activo de un compuesto que se forma cuando el compuesto se metaboliza. El término "metabolizado", como se usa en este documento, se refiere a la suma de los procesos (que incluyen, pero no se limitan a, reacciones de hidrólisis y reacciones catalizadas por enzimas) mediante los cuales un organismo cambia una sustancia particular. Por tanto, las enzimas pueden producir alteraciones estructurales específicas en un compuesto. Después de entrar en el cuerpo, la mayoría de los medicamentos son sustratos para reacciones químicas que pueden cambiar sus propiedades físicas y efectos biológicos. Estas conversiones metabólicas, que suelen afectar a la polaridad de los compuestos de la invención, alteran la forma en que los fármacos e distribuyen y excretan en el organismo. Sin embargo, en algunos casos, se requiere el metabolismo de un fármaco para obtener un efecto terapéutico. La invención también se refiere a profármacos de los compuestos para uso según la invención. El término "prodroga" o "profármaco" tal como aquí se utiliza incluye cualquier derivado de un compuesto de fórmula (I) , -por ejemplo y no limitativamente: ésteres (incluyendo ésteres de ácidos carboxílicos, ésteres de aminoácidos, ésteres de fosfato, ésteres de sulfonato de sales metálicas, etc.) , carbamatos, amidas, etc.- que al ser administrado a un individuo puede ser transformado directa o indirectamente en dicho compuesto de fórmula (I) en el mencionado individuo. Ventajosamente, dicho derivado es un compuesto que aumenta la biodisponibilidad del compuesto de fórmula (I) cuando se administra a un individuo o que potencia la liberación del compuesto de fórmula (I) en un compartimento biológico. La naturaleza de dicho derivado no es crítica siempre y cuando pueda ser administrado a un individuo y proporcione el compuesto de fórmula (I) , en un compartimento biológico de un individuo. La preparación de dicho profármaco puede llevarse a cabo mediante métodos convencionales conocidos por los expertos en la materia, que son capaces de liberar el compuesto de fórmula (I) como ingrediente activo cuando el profármaco se administra a un sujeto mamífero. La liberación del ingrediente activo ocurre in vivo. Los profármacos se pueden preparar mediante técnicas conocidas por los expertos en la técnica. Estas técnicas generalmente modifican los grupos funcionales apropiados en un compuesto dado. Sin embargo, estos grupos funcionales modificados regeneran grupos funcionales originales mediante manipulación rutinaria o in vivo. Los profármacos de los compuestos de la invención incluyen compuestos en los que se modifica un grupo hidroxi, amino, carboxílico o similar. Los ejemplos de profármacos incluyen, pero no se limitan a, ésteres. Los compuestos de fórmula general (I) y sus profármacos que contienen uno o varios centros quirales pueden estar presentes como racematos, mezclas diastereoméricas o isómeros individuales ópticamente activos. Los compuestos de la presente invención representados por la fórmula (I) pueden incluir isómeros, dependiendo de la presencia de enlaces múltiples, incluyendo isómeros ópticos o enantiómeros, dependiendo de la presencia de centros quirales. Los isómeros, enantiómeros o diastereoisómeros individuales y las mezclas de los mismos caen dentro del alcance de la presente invención, es decir, el término isómero también se refiere a cualquier mezcla de isómeros, como diastereómeros, racémicos, etc., incluso a sus isómeros ópticamente activos o las mezclas en distintas proporciones de los mismos. Los enantiómeros o diastereoisómeros individuales, así como sus mezclas, pueden separarse mediante técnicas convencionales. Composiciones y composiciones farmacéuticas El compuesto de fórmula (I) de la invención puede estar formando parte de una composición, preferiblemente de una composición farmacéutica. Otro aspecto de la invención se refiere a una composición que comprende el compuesto de fórmula (I) de la invención, de ahora en adelante composición de la invención, o una sal, estereoisómero, tautómero, hidrato o solvato farmacéuticamente aceptable del mismo, para la prevención y tratamiento del cáncer colorrectal. En una realización preferida de este aspecto de la invención, el cáncer colorrectal es el cáncer colorrectal metastásico. Más preferiblemente, donde el individuo presenta los niveles de hepsina elevados con respecto a los niveles normales. Otro aspecto de la invención se refiere al compuesto de la invención para la prevención o tratamiento de los eventos tromboembólicos venosos (ETEV) , y preferiblemente de aquellos desarrollados como consecuencia de la terapia antineoplásica. En otra realización de este aspecto, la composición de la invención además comprende otro principio activo. Más preferiblemente es una composición farmacéutica. En otra realización preferida además comprende vehículos farmacéuticamente aceptables. En otra realización preferida de este aspecto, la primera composición de la invención solo comprende como principio activo el compuesto de fórmula (I) , aunque puede comprender, además, excipientes y vehículos farmacéuticamente aceptables. El término "medicamento", tal y como se usa en esta memoria, hace referencia a cualquier sustancia usada para prevención, diagnóstico, alivio, tratamiento o curación de enfermedades en el hombre y los animales. La administración de los compuestos, composiciones o formas farmacéuticas de la presente invención puede ser realizada mediante cualquier método adecuado, como la infusión intravenosa y las vías oral, tópica o parenteral. La administración oral es la preferida por la conveniencia de los pacientes. La cantidad administrada de un compuesto de la presente invención dependerá de la relativa eficacia del compuesto elegido, la severidad de la enfermedad a tratar y el peso del paciente. Sin embargo, los compuestos de esta invención serán administrados una o más veces al día, por ejemplo 1, 2, 3 O 4 veces diarias, con una dosis total entre 0.1 y 1000 mg/Kg/día. Es importante tener en cuenta que puede ser necesario introducir variaciones en la dosis, dependiendo de la edad y de la condición del paciente, así como modificaciones en la vía de administración. "Composición farmacéutica", como se usa en el presente documento, se refiere a composiciones y entidades moleculares que son fisiológicamente tolerables y no producen típicamente una reacción alérgica o una reacción desfavorable similar a como trastornos gástricos, mareos y similares, cuando se administra a un humano o animal. Preferiblemente, el término "farmacéuticamente aceptable" significa que está aprobado por una agencia reguladora de un gobierno estatal o federal o está incluido en la Farmacopea de los Estados Unidos u otra farmacopea generalmente reconocida para uso en animales, y más particularmente en humanos. El término "excipiente" se refiere a un vehículo, diluyente o adyuvante que se administra con el ingrediente activo. Dichos excipientes farmacéuticos pueden ser líquidos estériles, como agua y aceites, incluidos los de origen petrolífero, animal, vegetal o sintético, como aceite de cacahuete, aceite de soja, aceite mineral, aceite de sésamo y similares. Como vehículos se utilizan preferiblemente agua o soluciones acuosas salinas y soluciones acuosas de dextrosa y glicerol, particularmente para soluciones inyectables. Los vehículos farmacéuticos adecuados se describen en "Remington's Phamaceutical Sciences" de EW Martin, 21a edición, 2005; o "Manual de excipientes farmacéuticos", Rowe CR; Paul JS; Marian EQ, sexta edición. Como se emplea aquí, el término "principio activo", "substancia activa", "substancia farmacéuticamente activa", "ingrediente activo" ó "ingrediente farmacéuticamente activo" significa cualquier componente que potencialmente proporcione una actividad farmacológica u otro efecto diferente en el diagnóstico, cura, mitigación, tratamiento, o prevención de una enfermedad, o que afecta a la estructura o función del cuerpo del hombre u otros animales. El término incluye aquellos componentes que promueven un cambio químico en la elaboración del fármaco y están presentes en el mismo de una forma modificada prevista que proporciona la actividad específica o el efecto. Las cantidades apropiadas de un compuesto de fórmula (I) como se define anteriormente, o una sal, estereoisómero farmacéuticamente aceptable o solvato del mismo se pueden formular con excipientes y/o vehículos farmacéuticamente aceptables para obtener una composición farmacéutica para uso en los usos médicos de la invención. Los vehículos farmacéuticamente aceptables adecuados incluyen, por ejemplo, agua, soluciones salinas, alcohol, aceites vegetales, polietilenglicoles, gelatina, lactosa, amilosa, estearato de magnesio, talco, tensioactivos, ácido silícico, parafina viscosa, aceite perfumado, monoglicéridos y diglicéridos. de ácidos grasos, ésteres de ácidos grasos, petroetrales, hidroximetilcelulosa, polivinilpirrolidona y similares. Las composiciones farmacéuticas que contienen el compuesto de fórmula (I) , como se definió anteriormente, o una sal, estereoisómero o solvato farmacéuticamente aceptable del mismo para su uso de acuerdo con la invención pueden ocurrir en cualquier forma farmacéutica de administración considerada apropiada para la vía de administración seleccionada, por ejemplo, por administración sistémica (por ejemplo, inyección intravenosa, subcutánea, intramuscular) , oral, parenteral o tópica, para lo cual incluirá los excipientes farmacéuticamente aceptables necesarios para la formulación del método de administración deseado. Además, también es posible administrar la composición que comprende el compuesto de fórmula (I) como se definió anteriormente, o una sal farmacéuticamente aceptable, estereoisómero o solvato del mismo para uso según la invención por vía intranasal o sublingual que permite la administración sistémica mediante un modo de administración no agresivo. Además, la administración intraventricular puede ser adecuada. Una vía de administración preferida es la oral. Cuando sea necesario, el compuesto de fórmula (I) como se definió anteriormente, o una sal, estereoisómero o solvato farmacéuticamente aceptable del mismo para su uso de acuerdo con la invención está comprendido en una composición que también incluye un agente solubilizante y un anestésico local para aliviar cualquier dolor en el lugar de la inyección. Generalmente, los ingredientes se suministran por separado o mezclados en forma de dosificación unitaria, por ejemplo, como un polvo liofilizado seco o un concentrado libre de agua en un recipiente herméticamente cerrado como una ampolla o bolsita que indica la cantidad de agente activo. Cuando la composición deba administrarse mediante infusión, se puede dispensar con una botella de infusión que contenga agua o solución salina estériles de calidad farmacéutica. En casos distintos de la administración intravenosa, la composición puede contener cantidades menores de agentes humectantes o emulsionantes, o agentes tamponantes del pH. La composición puede ser una solución líquida, suspensión, emulsión, gel, polímero o formulación de liberación sostenida. La composición se puede formular con aglutinantes y vehículos tradicionales, como se conoce en la técnica. Las formulaciones pueden incluir vehículos estándar tales como grados farmacéuticos de manitol, lactosa, almidón, estearato de magnesio, sacárido de sodio, celulosa, carbonato de magnesio, etc., vehículos inertes que tienen una funcionalidad bien establecida en la fabricación de productos farmacéuticos. Se conocen varios sistemas de administración y se pueden usar para administrar un compuesto de la presente invención que incluye la encapsulación en liposomas, micropartículas, microcápsulas y similares. Otro aspecto de la invención se refiere a una forma farmacéutica, de ahora en adelante forma farmacéutica de la invención, que comprende un compuesto de la invención (un compuesto de fórmula (I) , o la composición de la invención. En esta memoria se entiende por "forma farmacéutica" la mezcla de uno o más principios activos con o sin aditivos que presentan características físicas para su adecuada dosificación, conservación, administración y biodisponibilidad. Las formas de dosificación sólidas para administración oral pueden incluir cápsulas convencionales, cápsulas de liberación sostenida, comprimidos convencionales, comprimidos, comprimidos masticables, comprimidos sublinguales, efervescentes comprimidos, píldoras, suspensiones, polvos, gránulos y geles de liberación sostenida. En estas formas de dosificación sólidas, los compuestos activos se pueden mezclar con al menos un excipiente inerte como sacarosa, lactosa o almidón. Tales formas de dosificación también pueden comprender, como en la práctica normal, sustancias adicionales distintas de diluyentes inertes, por ejemplo, agentes lubricantes tales como estearato de magnesio. En el caso de cápsulas, comprimidos, comprimidos efervescentes y píldoras, las formas de dosificación también pueden comprender agentes tamponantes. Los comprimidos y las píldoras se pueden preparar con recubrimientos entéricos. Las formas de dosificación líquidas para administración oral pueden incluir emulsiones, soluciones, suspensiones, jarabes y elixires farmacéuticamente aceptables que contienen diluyentes inertes usados comúnmente en la técnica, tales como agua. Esas composiciones también pueden comprender adyuvantes tales como agentes humectantes, agentes mulsionantes y de suspensión y agentes edulcorantes, agentes aromatizantes y perfumantes. Las preparaciones inyectables, por ejemplo, suspensiones acuosas u oleaginosas, inyectables estériles se pueden formular de acuerdo con la técnica conocida usando agentes dispersantes, agentes humectantes y/o agentes de suspensión adecuados. Entre los vehículos y disolventes aceptables que se pueden utilizar se encuentran el agua, la solución de Ringer y la solución isotónica de cloruro de sodio. Los aceites estériles también se utilizan convencionalmente como disolventes o medios de suspensión. Para la administración tópica, los compuestos de la invención pueden formularse como cremas, geles, lociones, líquidos, pomadas, rocíe soluciones, dispersiones, barras sólidas, emulsiones, microemulsiones y similares que se pueden formular de acuerdo con métodos convencionales que utilizan excipientes adecuados, tales como, por ejemplo, emulsionantes, tensioactivos, agentes espesantes, agentes colorantes y combinaciones de dos o más de los mismos. Adicionalmente, el compuesto de fórmula (I) como se definió anteriormente, o una sal farmacéuticamente aceptable, estereoisómero o solvato del mismo para uso según la invención se pueden administrar en forma de parches transdérmicos o iontoforesis dispositivos. En una realización, los compuestos para uso según la invención se administran como un parche transdérmico, por ejemplo, en forma de parche transdérmico de liberación sostenida. Se conocen en la técnica parches transdérmicos adecuados. Varios sistemas de suministro de fármacos son conocidos y se pueden utilizar para administrar los agentes o composiciones para uso de acuerdo con la invención, incluyendo, por ejemplo, encapsulación en liposomas, microburbujas, emulsiones, micropartículas, microcápsulas y similares. La dosis requerida se puede administrar como una sola unidad o en una forma de liberación sostenida. Las formas de liberación sostenible y los materiales y métodos apropiados para su preparación se describen, por ejemplo, en " Modified-Release Drug Deliver y Technology ", Rathbone, MJ Hadgraft, J. y Roberts, MS (eds.) , Marcel Dekker, Inc., Nueva York (2002) , " Handbook of Pharmaceutical Controlled Release Technology ", Wise, DL (ed.) , Marcel Dekker, Inc. Nueva York, (2000) . En una realización de la invención, la forma administrable por vía oral de un compuesto para uso según la invención está en una forma de liberación sostenida ue comprende además al menos un recubrimiento o matriz. El recubrimiento o matriz de liberación sostenida incluye, sin limitación, polímeros naturales, ceras, grasas, semisintéticos o sintéticos insolubles en agua, modificados, alcoholes grasos, ácidos grasos, plastificantes naturales semisintéticos o sintéticos, o una combinación de dos o más de ellos. Los recubrimientos entéricos se pueden aplicar usando procesos convencionales conocidos por los expertos en la técnica, como se describe en, por ejemplo, Johnson, JL, " Pharmaceutical tablet coating", Coatings Technology Handbook (Segunda edición) , Satas, D. y Tracton, AA (eds. ) , Marcel Dekker, Inc. Nueva York, (2001) , Carstensen, T., " Coating Tablets in Advanced Pharmaceutical Solids", Swarbrick, J. (ed.) , Marcel Dekker, Inc. Nueva York (2001) , 455-468. El término "prevención", "prevenir" o "prevenir", como se usa en el presente documento, se refiere a la administración de un compuesto de acuerdo con la invención o de un medicamento que comprende dicho compuesto a un sujeto que no ha sido diagnosticado como posiblemente tener una enfermedad, pero que normalmente se esperaría que la desarrolle o que tenga un mayor riesgo de padecerla. La prevención pretende evitar la aparición de dicha enfermedad. La prevención puede ser completa (por ejemplo, la ausencia total de una enfermedad) . La prevención también puede ser parcial, de modo que, por ejemplo, la aparición de una enfermedad en un sujeto es menor que la que habría ocurrido sin la administración del compuesto de la presente invención. La prevención también se refiere a la reducción de la susceptibilidad a una condición clínica. El término "tratamiento", como se usa en este documento, se refiere a cualquier tipo de terapia, que tiene como objetivo terminar, prevenir, mejorar o reducir la susceptibilidad a una condición clínica como se describe en este documento. En una realización preferida, el término tratamiento se refiere a un tratamiento profiláctico (es decir, una terapia para reducir la susceptibilidad a una afección clínica) , de un trastorno o afección como se define en el presente documento. Por tanto, "tratamiento", "tratar" y sus términos equivalentes se refieren a la obtención de un efecto farmacológico o fisiológico deseado, que cubre cualquier tratamiento de una afección o trastorno patológico en un mamífero, incluido un ser humano. El efecto puede ser profiláctico en términos de prevención total o parcial de un trastorno o síntoma del mismo y / o puede ser terapéutico en términos de una cura parcial o completa de un trastorno y / o efecto adverso atribuible al trastorno. Es decir, "tratamiento" incluye (1) evitar que el trastorno ocurra o reaparezca en un sujeto, (2) inhibir el trastorno, como detener su desarrollo, (3) detener o terminar el trastorno o, al menos, los síntomas asociados con el mismo, de modo que el huésped ya no sufra el trastorno o sus síntomas, como provocar la regresión del trastorno o sus síntomas, por ejemplo, al restaurar o reparar una función perdida, altante o defectuosa, o estimular un proceso ineficiente, o (4) aliviar, aliviar o mejorar el trastorno o los síntomas asociados con el mismo. El término "sujeto" como se usa en el presente documento, se refiere a cualquier sujeto, particularmente un sujeto mamífero, para el que se desea la terapia. Los sujetos mamíferos incluyen humanos, animales domésticos, animales de granja y zoológicos, deportes o animales de compañía como perros, gatos, cobayas, conejos, ratas, ratones, caballos, ganado, vacas, etc. En una realización preferida de la invención, el sujeto es un mamífero. En una realización más preferida de la invención, el sujeto es un ser humano. La administración de los compuestos, composiciones o formas farmacéuticas de la presente invención puede ser realizada mediante cualquier método adecuado, como la infusión intravenosa y las vías oral, tópica o parenteral. La administración oral es la preferida por la conveniencia de los pacientes. La cantidad administrada de un compuesto de la presente invención dependerá de la relativa eficacia del compuesto elegido, la severidad de la enfermedad a tratar y el peso del paciente. Sin embargo, los compuestos de esta invención serán administrados una o más veces al día, por ejemplo 1, 2, 3 ó 4 veces diarias, con una dosis total entre 0.1 y 1000 mg/Kg/día. Es importante tener en cuenta que puede ser necesario introducir variaciones en la dosis, dependiendo de la edad y de la condición del paciente, así como modificaciones en la vía de administración. A lo largo de la descripción y las reivindicaciones la palabra "comprende" y sus variantes no pretenden excluir otras características técnicas, aditivos, componentes o pasos. Para los expertos en la materia, otros objetos, ventajas y características de la invención se desprenderán en parte de la descripción y en parte de la práctica de la invención. Los siguientes ejemplos y figuras se proporcionan a modo de ilustración, y no se pretende que sean limitativos de la presente invención. EJEMPLOS DE LA INVENCIÓN Cribado virtual Con el objetivo de encontrar nuevos inhibidores para la hepsina se realizó un cribado virtual mediante la técnica de docking molecular a partir de la estructura cristalográfica de la proteína epositada en la Protein Data Bank con código 1P57 frente a la librería de compuestos DrugBank (https://go.drugbank.com/) en las coordenadas de su sitio catalítico, caracterizada por residuos HIS57, ASP102 y SER195. El docking molecular se realizó en un clúster de cónputo de alto rendimiento mediante el software Autodock Vina 1.1.2 (http://vina.scripps.edu/) y para ello se convirtieron tanto la estructura de la proteína como la librería de ligandos DrugBank al formato pdbqt mediante MGLTools. Una vez terminados los cálculos se priorizaron los compuestos en función de docking score y se examinaron visualmente los 8 primeros, seleccionando el compuesto Suramin (código DrugBank DB04786 y primero en la lista) con un docking score de -12 Kcal/mol e interaciones hidrofóbicas con los residuos ALA-39, LEU-41, HIS-57, PRO-60, PHE-97, GLN-151, interacciónes mediante puente de hidrogeno con los residuos GLY-38, HIS-40, HIS-57, ASN-63, GLN-73, ASN-99, GLN-151, GLY-153, SER-195, SER-214, GLY-216, GLY-219, interacciones por "puentes de sal" con ARG-34, HIS-57 y una interacción "piStacking" con el residuo HIS-57 como se aprecia en la Figura 1. En dicha figura podemos observar que Suramin impide el acceso a la triada catalítica, representada en formato de superficie para sus tres residuos Suramin actúa como inhibidor irreversible de hepsina. Mediante un ensayo fluorogénico se determinó la capacidad de Suramin de inhibir la actividad proteolítica de hepsina. Hepsina es una serín proteasas capaz de hidrolizar al sustrato BOC-Gln-Arg-Arg-AMC (Bachem, Barcelona, Spain) , de manera que es posible registrar la emisión de fluorescencia mediante un lector de placas con longitudes de onda de excitación y emisión de 380 nm y 460 nm, respectivamente. Para ello, se incubó hepsina (0.05 j M) (R&D Systems, Madrid, Spain) en tampón 50 mM Tris-HCl, pH 9 buffer, con 200 j M BOC-Gln-Arg-Arg-AMC y se registró la fluorescencia emitida durante 5 minutos. Para comprobar el efecto de Suramin, se llevó a cabo la misma reacción, pero incubando previamente hepsina con Suramin durante 1 hora a 37°C y en distintas concentraciones que oscilaron entre 0.13 y 10 |jM. De esta forma se calculó el valor de IC50, o concentración de inhibidor a la que se alcanza la mitad de la velocidad máxima de hepsina en la hidrólisis de su sustrato, que fue de 0.66 j M. Suramin reduce la migración de células de cáncer colorrectal Una vez que las células alcanzan confluencia, forman una monocapa. Mediante la eliminación de una tira de células 300-500 ^m de anchura con una punta de pipetea de 200 ^l, es posible evaluar la capacidad de migración de las células y el efecto de Suramín en este proceso. La capacidad de migración se evalúa en función del porcentaje de área ocupada tras 48 horas de incubación y se cuantifica utilizando el software ImageJ. Los resultados muestran que Suramín a una concentración de 0, 66 ^M reduce de forma significativa la migración de células Caco-2, tanto con expresión basal como con sobreexpresión de hepsina, mediante transfección estable de un plásmido que contiene el gen HPN. Suramin reduce la invasión de células de cáncer colorrectal Para evaluar la invasión se realizó un ensayo en el que se evaluó la capacidad de las células para degradar una matriz de gelatina. Para ello, se mezcló gelatina al 0.2% y rodamina (Invitrogen, Life Technologies, Madrid, Spain) , en una ratio 1:55 en tampón NaCl 61 mM, borohidrato sódico 50 mM y después se dializó toda la noche en PBS. Se prepararon cubres con la mezcla preparada cubriéndolos y se fijó la matriz con glutaraldehido 0.5% durante 15 min. Los cubres se lavaron con PBS y se añadieron las suspensiones de células Caco-2 durante 72 horas. Posteriormente se fijaron las células con formaldehído 3.7% y se incubó con phalloidin faloidin?? (0, 01 mg/ml) y ProLongGold Antifade con DAPI (4', 6-diamino-2-phenylindole; ThermoFisher) . Las imágenes se tomaron con un microscopio confocal SP8 LEICA y se analizaron con ImageJ. Los resultados mostraron que Suramín, a una concentración de 0, 66 ^M, reduce de forma significativa la degradación de la gelatina de células Caco-2 tanto con expresión basal como con sobreexpresión de hepsina. Suramin reduce la generación de trombina Puesto que hepsina activa al FVII y, por tanto, la cascada de la coagulación, podría contribuir al estado de hipercoagulabilidad de los pacientes con cáncer colorrectal con niveles elevados de hepsina. Para comprobar la hipótesis, se incubó plasma procedente de 20 sujetos sanos, a los que se extrajo sangre citratada 3.8% mediante venopunción, con las células Caco-2 con sobreexpresión de hepsina. El plasma se centrifugó previamente a 2500 g 20 minutos. Para el ensayo de generación de trombina, se retiró el plasma incubado y se incubó con el reactivo LOW® (factor tisular: 1 pmol; fosfolípidos: 4 ^mol; Diagnostica Stago) en una placa de 96 pocillo. Todas las muestras se prepararon en duplicado. La coagulación en estas muestras se inició añadiendo cloruro cálcico en un tampón que contenía el sustrato fluorogénico FluCa-kit reagent® (Diagnostica Stago) . Para cada muestra individual se utilizó un calibrador de rombina. La fluorescencia se registró durante 60 min en un fluorímetro Fluoroskan Ascent (Thermolab Systems) y los datos se analizaron utilizando el software Thrombinoscope™ (version 5.0.0.742; Diagnostica Stago) . Se analizaron los siguientes parámetros: (a) lag-time, que indica el inicio de la fase de generación de trombina; (b) tiempo hasta alcanzar la máxima concentración de trombina (ttPeak) ; (c) concentración máxima de trombina (Peak) ; (d) índice de frecuencia media (MRI) de la fase de propagación de la generación de trombina calculada por la fórmula Peak/ (ttPeak - lag-time) y se expresa en nM/min; y (e) potencial endógeno de trombina (ETP) que muestra la actividad enzimática de trombina evaluada como el área bajo la curva. Los resultados muestran que Suramín reduce el potencial endógeno de trombina (ETP) , el pico máximo (peak) y la velocidad en alcanzar el pico mientras que provoca un incremento en el tiempo de inicio de la fase de generación de trombina (lag-time) y el tiempo en alcanzar el pico. Tabla 1. Valores promedios del test de generación de trombina. Suramin no afecta ni a la muerte celular, ni a la proliferación celular Se quiso evaluar si la menor migración o invasión celular podría deberse a una mayor muerte, o a una menor proliferación celular. Para ello se incubaron las células (5x105 cells/ml) en placas de 6 pocillos en presencia o ausencia de Suramín tras 48 h. El tipo de muerte celular se determinó mediante el ensayo de anexina V/7-ADD (Anexin V Apoptosis Detection Kit FITC; eBiosciences, Thermo Fisher, Karlsruhe, Alemania; 7-ADD BD-Biosciences) siguiendo las instrucciones del fabricante. Para evaluar la actividad proliferativa de las células Caco-2, se añadió 5-etinil-2'-desoxiuridina (EdU) (Thermo Fisher Scientific) durante 48 h. Luego, se usaron alícuotas de las células tumorales para determinar el porcentaje de células positivas, es decir, Caco-2 o Caco-2-HPN-EdU+, detectadas por reacción de acoplamiento de azida luorescente con EdU de acuerdo con el protocolo del fabricante (Click-iT; Thermo Fisher Scientific) . Los resultados mostraron que Suramín no afecta ni a la muerte (porcentajes entorno al 5-10 %) ni a la proliferación celular. Suramin inhibe la invasión de las células de cáncer colorrectal en modelos de xenotrasplante de larvas de pez cebra Se obtuvieron peces cebra wild type (Danio rerio H. Cypriniformes, Cyprinidae) del Zebrafish International Resource Center (ZIRC, Oregon, EE. UU.) Los peces roya9/a9; nacrew2/w2 (casper) transparentes se obtuvieron como ya se ha descrito previamente en la bibliografía [R.M. White, A. Sessa, C. Burke, T. Bowman, J. LeBlanc, C. Ceol, et al. Transparent adult zebrafish as a tool for in vivo transplantation analysis. Cell Stem Cell 2008;2:183-9]. Los peces se aparearon, clasificaron, criaron y procesaron como se describe en el manual del pez cebra [M. Westerfield. The zebrafish book: a guide for the laborator y use of zebrafish. Http//Zfin Org/Zf_info/Zfbook/Zfbk Html 2000.]. Los huevos de pez cebra fertilizados se obtuvieron del desove natural y se mantuvieron en nuestras instalaciones siguiendo las prácticas estándar de cría. Los animales se mantuvieron en un ciclo de luz / oscuridad de 12 h a 28 ° C. Las larvas de pez cebra se anestesiaron con 0.16 mg/ml ethyl 3-aminobenzoato (tricaine; Sigma-Aldrich) . Las células Caco-2 y Caco-2-HPN se cultivaron en presencia y ausencia de 0, 66 ^M de suramin durante 48 h, luego se disgregaron y marcaron con 1, 1'-di-octa-decil-3, 3, 3 ', 3'-perclorato de tetra-metil-indo-carbo-cianina (Dil, ThermoFisher) y finalmente se resuspendieron en un tampón que contenía suero bovino fetal al 5% en PBS. La inyección de las células en embriones se realizó mediante la inyección de 200 células/embrión en el saco vitelino de larvas de pez cebra wild type o Casper 48 horas después de la fertilización y después de 5 días a 35°C, las larvas se analizaron mediante microscopía de fluorescencia para comprobar la diseminación de las células tumorales. La puntuación de la invasión de células Caco-2 y Caco-2-HPN se calculó como el porcentaje de larvas con invasión de células tumorales con respecto al total de larvas analizadas. El pretratamiento de las células Caco-2-HPN (CACO HPN) con suramin (HPN + S) redujo su capacidad de invasión a los niveles encontrados en las células parentales (CACO WT) (Figura 8) , lo que confirma los estudios in vitro y muestra además su potencial terapéutico para el tratamiento del cáncer colorrectal.
Publicaciones:
ES2912350 (25/05/2022) - A1 Solicitud de patente con informe sobre el estado de la técnica
ES2912350 (13/11/2023) - B2 Patente de invención con examen
ES2912350 (05/12/2023) - B9 Patente de invención corregida
Eventos:
En fecha 19/09/2021 se realizó Registro Instancia de Solicitud
En fecha 19/09/2021 se realizó Admisión a Trámite
En fecha 19/09/2021 se realizó Aceptación Tramitación CAP
En fecha 19/09/2021 se realizó 1001P_Comunicación Admisión a Trámite
En fecha 22/09/2021 se realizó Superado examen de oficio
En fecha 13/05/2022 se realizó Realizado IET
En fecha 17/05/2022 se realizó 1109P_Comunicación Traslado del IET
En fecha 25/05/2022 se realizó Publicación Solicitud
En fecha 25/05/2022 se realizó Publicación Folleto Solicitud con IET (A1)
En fecha 24/08/2022 se realizó PETEX_Petición de examen sustantivo
En fecha 09/09/2022 se realizó Validación petición y/o pago de examen sustantivo conforme
En fecha 29/11/2022 se realizó El solicitante ha contestado pero existen nuevas objeciones a la concesión de la solicitud
En fecha 29/11/2022 se realizó Elaboración de examen sustantivo
En fecha 30/11/2022 se realizó 6120P_Notificación de examen sustantivo
En fecha 05/12/2022 se realizó Publicación de examen sustantivo
En fecha 23/01/2023 se realizó No Inscripcion de Cesion F202230730
En fecha 06/02/2023 se realizó 5127P_Subsanación a defectos en examen sustantivo
En fecha 30/03/2023 se realizó 6120P_Notificación de examen sustantivo
En fecha 05/04/2023 se realizó Publicación de examen sustantivo
En fecha 05/06/2023 se realizó 3585X_Registro Solicitud Prórroga de Plazos
En fecha 05/06/2023 se realizó Concesión Prórroga de Plazos
En fecha 05/06/2023 se realizó 1585X_Notificación Concesión Prórroga de Plazos
En fecha 09/06/2023 se realizó Publicación Concesión Prórroga de Plazos (BOPI)
En fecha 07/08/2023 se realizó 5127P_Subsanación a defectos en examen sustantivo
En fecha 19/10/2023 se realizó Designación de Comisión de Expertos
En fecha 20/10/2023 se realizó Finalización de Examen Sustantivo
En fecha 20/10/2023 se realizó 6121P_Comunicación finalización de examen sustantivo
En fecha 26/10/2023 se realizó Publicación finalización de examen sustantivo
En fecha 03/11/2023 se realizó Concesión con examen sustantivo
En fecha 03/11/2023 se realizó Entrega título
En fecha 03/11/2023 se realizó 6125P_Notificación de concesión con examen sustantivo
En fecha 13/11/2023 se realizó Publicación concesión Patente
En fecha 13/11/2023 se realizó Publicación Folleto Concesión
En fecha 05/12/2023 se realizó Publicación de la Patente de invención corregida (BOPI)
En fecha 05/12/2023 se realizó Publicación Folleto Patente deInvención Corregida (B9)
Pagos:
19/09/2021 - Pago Tasas IET
14/12/2023 - Pago 03 Anualidad
Fuente de la información
Parte de la información aquí publicada es pública puesto que ha sido obtenida de la Oficina de Propiedad Industrial de los diferentes países el 26/04/2024 y por lo tanto puede ser que la información no esté actualizada.Parte de la información aquí mostrada ha sido calculada por nuestro sistema informático y puede no ser veraz.
Privacidad
Si considera que al información aquí publicada afecta a su privacidad y desea que eliminemos la información aquí publicada envíe un email a info@patentes-y-marcas.com o rellene el formulario que encontrará aquí.Información sobre el registro de patente nacional por NUEVO TRATAMIENTO DEL CÁNCER COLORRECTAL con el número P202130871
El registro de patente nacional por NUEVO TRATAMIENTO DEL CÁNCER COLORRECTAL con el número P202130871 fue solicitada el 19/09/2021. Se trata de un registro en España por lo que este registro no ofrece protección en el resto de países. El registro NUEVO TRATAMIENTO DEL CÁNCER COLORRECTAL con el número P202130871 fue solicitada por FUNDACION UNIVERSITARIA SAN ANTONIO mediante los servicios del agente MARIA DESAMPARADOS DIAZ PACHECO. El registro [modality] por NUEVO TRATAMIENTO DEL CÁNCER COLORRECTAL con el número P202130871 está clasificado como A61K 31/185,A61P 35/00,A61P 35/04,A61P 7/02 según la clasificación internacional de patentes.
Otras invenciones solicitadas por FUNDACION UNIVERSITARIA SAN ANTONIO
Es posible conocer todas las invenciones solicitadas por FUNDACION UNIVERSITARIA SAN ANTONIO entre las que se encuentra el registro de patente nacional por NUEVO TRATAMIENTO DEL CÁNCER COLORRECTAL con el número P202130871. Si se desean conocer más invenciones solicitadas por FUNDACION UNIVERSITARIA SAN ANTONIO clicar aquí.Otras invenciones solicitadas en la clasificación internacional de patentes A61K 31/185,A61P 35/00,A61P 35/04,A61P 7/02.
Es posible conocer invenciones similares al campo de la técnica se refiere. El registro de patente nacional por NUEVO TRATAMIENTO DEL CÁNCER COLORRECTAL con el número P202130871 está clasificado con la clasificación A61K 31/185,A61P 35/00,A61P 35/04,A61P 7/02 por lo que si se desea conocer más registros con la clasificación A61K 31/185,A61P 35/00,A61P 35/04,A61P 7/02 clicar aquí.Otras invenciones solicitadas a través del representante MARIA DESAMPARADOS DIAZ PACHECO
Es posible conocer todas las invenciones solicitadas a través del agente MARIA DESAMPARADOS DIAZ PACHECO entre las que se encuentra el registro patente nacional por NUEVO TRATAMIENTO DEL CÁNCER COLORRECTAL con el número P202130871. Si se desean conocer más invenciones solicitadas a través del agente MARIA DESAMPARADOS DIAZ PACHECO clicar aquí.Patentes en España
Es posible conocer todas las invenciones publicadas en España entre las que se encuentra el registro patente nacional por NUEVO TRATAMIENTO DEL CÁNCER COLORRECTAL. Nuestro portal www.patentes-y-marcas.com ofrece acceso a las publicaciones de patentes en España. Conocer las patentes registradas en un país es importante para saber las posibilidades de fabricar, vender o explotar una invención en España.Patentes registradas en la clase A
Es posible conocer todas las patentes registradas en la clase A (NECESIDADES CORRIENTES DE LA VIDA) entre las que se encuentra la patente NUEVO TRATAMIENTO DEL CÁNCER COLORRECTAL con el número P202130871. Conocer las patentes registradas en una clase es importante para saber las posibilidades de registrar una patente en esa misma clase.Patentes registradas en la clase A61
Es posible conocer todas las patentes registradas en la clase A61 (CIENCIAS MEDICAS O VETERINARIAS; HIGIENE) entre las que se encuentra la patente NUEVO TRATAMIENTO DEL CÁNCER COLORRECTAL con el número P202130871. Conocer las patentes registradas en una clase es importante para saber las posibilidades de registrar una patente en esa misma clase.Patentes registradas en la clase A61K
Es posible conocer todas las patentes registradas en la clase A61K (PREPARACIONES DE USO MEDICO, DENTAL O PARA EL ASEO ) entre las que se encuentra la patente NUEVO TRATAMIENTO DEL CÁNCER COLORRECTAL con el número P202130871. Conocer las patentes registradas en una clase es importante para saber las posibilidades de registrar una patente en esa misma clase.Patentes registradas en la clase A61P
Es posible conocer todas las patentes registradas en la clase A61P (ACTIVIDAD TERAPEUTICA ESPECIFICA DE COMPUESTOS QUIMICOS O DE PREPARACIONES MEDICINALES) entre las que se encuentra la patente NUEVO TRATAMIENTO DEL CÁNCER COLORRECTAL con el número P202130871. Conocer las patentes registradas en una clase es importante para saber las posibilidades de registrar una patente en esa misma clase.
¿Tienes alguna duda?
Escribe tu consulta y te responderemos rápida y gratuitamente.

Otras patentes similares
 P202130621
P202130621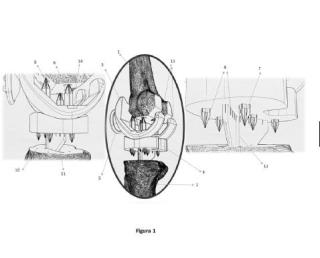 P202130869
P202130869 P202130985
P202130985Profesionales Recomendados
Barcelona
933182440
España
933182440
España

Barcelona
+34 93 362 16 97
España
+34 93 362 16 97
España

Barcelona
932 593 600
España
932 593 600
España







