


- Home /
- Publicaciones de patentes /
- IMPLANTE CRANEOFACIAL DE POLIÉTER ÉTER CETONA (PEEK) CON DEPÓSITOS PARA ALMACENAR Y LIBERAR SUSTANCIAS ACTIVAS
Patente nacional por "IMPLANTE CRANEOFACIAL DE POLIÉTER ÉTER CETONA (PEEK) CON DEPÓSITOS PARA ALMACENAR Y LIBERAR SUSTANCIAS ACTIVAS"
Este registro ha sido solicitado por
Persona física
a través del representanteNURIA CAPITAN GARCÍA
Contacto
- Estado: Vigente
- País:
- España
- Fecha solicitud:
- 20/01/2023
- Número solicitud:
-
P202330037
- Número publicación:
-
ES2957984
- Fecha de concesión:
-
- Inventores:
-
Persona física
- Datos del titular:
-
Persona física
- Datos del representante:
-
Nuria Capitan García
- Clasificación Internacional de Patentes:
- A61F 2/28
- Clasificación Internacional de Patentes de la publicación:
- A61F 2/28
- Fecha de vencimiento:
Quiero registrar una patente

Reivindicaciones:
+ ES-2957984_A11. Implante craneofacial de material PEEK para la liberación de sustancias o principios activos caracterizado porque comprende: Uno o más depósitos o recámaras interiores (1) ovaladas con paredes de 2, 5 a 4 mm de grosor, que almacenan un líquido o sustancia activa en formato líquido para ser liberado de forma gradual y prolongada al sitio del implante, tuberías (2) que interconectan los depósitos o recámaras interiores con los orificios de entrada y salida, definiéndose: a) una tubería de entrada que conecta el orifico de entrada (3) con el depósito (1) de tal forma que incluye un sistema de embudo que incluye una entrada o carga del líquido que tiene un diámetro de 1, 5-2, 0 mm y que luego se ensancha en una tubería de diámetro 2, 0 a 3, 0 mm para continuar con la tubería hasta el depósito. b) una tubería de salida que conecta el depósito o recámara (1) con el orificio de salida (4) que incluye un sistema de embudo de transición que presenta un diámetro de 07 a 2, 0 mm en la parte superior y un diámetro de 0, 7 a 1, 5 mm en su parte inferior, dependiendo del diámetro del orificio de salida (4) , Uno o más orificios de entrada (3) en la parte superior del implante dependiendo del número de depósitos, donde dicho orificio es la entrada o carga del líquido para llegar al o los depósitos (1) , donde el orificio de entrada tiene un diámetro de 1, 5-2, 0 mm y está bloqueado por un tapón de filamento PEEK o un tornillo de titanio, Uno o más orificios de salida (4) en la parte inferior del implante dependiendo del número de depósitos como orificio de salida gradual y prolongada del líquido o sustancia activa hacia el sitio del implante, donde el orificio de salida tiene un diámetro de 0, 7 a 1, 5mm y tiene un tapón o sistema que sella el implante para no permitir que el líquido se escape cuando no se ha dispuesto en el sitio del implante. 2. Implante craneofacial de material PEEK para la liberación de sustancias o principios activos de acuerdo con la reivindicación 1 caracterizado porque el orificio de entrada tiene un diámetro de 1, 5 mm. 3. Implante craneofacial de material PEEK para la liberación de sustancias o principios activos de acuerdo con la reivindicación 1 caracterizado porque el orificio de salida del líquido o sustancia activa tiene un diámetro de 0, 75mm. 4. Implante craneofacial de material PEEK para la liberación de sustancias o principios activos de acuerdo con la reivindicación 1 caracterizado porque el grosor de las paredes de los depósitos o recámaras es de 3, 5 mm. 5. Implante craneofacial de material PEEK para la liberación de sustancias o principios activos de acuerdo con la reivindicación 1 caracterizado porque las tuberías de todo el sistema tanto de entrada como de salida deben de ser de al menos 10 mm, con un máximo de 15 mm. 6. Uso del implante craneofacial de material PEEK para la liberación de sustancias o principios activos de acuerdo con las reivindicaciones 1 a 5 caracterizado porque sirve para la liberación prolongada y sostenida de sustancias y principios activos en formato líquido. 7. Uso del implante craneofacial de material PEEK para la liberación de sustancias o principios activos de acuerdo con la reivindicación 6 caracterizado porque sirve para la liberación prolongada y sostenida de al menos un principio activo soluble en un vehículo farmacéuticamente aceptable. 8. Uso del implante craneofacial de material PEEK para la liberación de sustancias o principios activos de acuerdo con la reivindicación 7 caracterizado porque sirve para la liberación prolongada y sostenida de principios activos analgésicos, antibióticos, antivirales, quimioterapéuticos, antiinflamatorios p cualquier principio activo soluble en un vehículo farmacéuticamente aceptable.
Los productos y servicios protegidos por este registro son:
A61F 2/28
Descripciones:
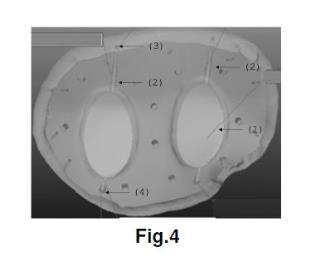
+ ES-2957984_A1 IMPLANTE CRANEOFACIAL DE POLIÉTER ÉTER CETONA (PEEK) CON DEPÓSITOS PARA ALMACENAR Y LIBERAR SUSTANCIAS ACTIVAS CAMPO DE LA INVENCIÓN La presente invención se relaciona con el área la biomedicina. Particularmente, se refiere a un implante craneofacial de material Poliéter éter cetona (PEEK) que dispone de depósitos para almacenar y liberar sustancias o principios activos en el sitio del implante. ANTECEDENTES DE LA INVENCIÓN En procedimientos quirúrgicos las infecciones son condiciones que con frecuencia son emergencias y que pueden determinar el fracaso o el éxito del procedimiento. En cirugías donde el procedimiento comprende la incorporación de prótesis, las complicaciones perioperatorias siguen siendo una preocupación importante. Una de las complicaciones más comunes y complejas son las infecciones bacterianas que pueden dar lugar a retrasos en la cicatrización, además de un aumento en los costos para el paciente e incluso la muerte (Chengzhe et al, 2021) . Este tipo de infecciones representan entre el 15 al 30% de las infecciones intrahospitalarias con una tasa de mortalidad entre 0, 6 y 1, 9% (Hernández et al., 2020) . La incidencia de infecciones posquirúrgicas depende del sitio y varía de <1 a 2% para reemplazos articulares y hasta 10% para cirugías de columna, además, estas tasas dependen de la salud del paciente y aumentan en pacientes de riesgo (ancianos, diabéticos e inmunodeprimidos) (Delaney et al., 2019) . En Chile, las infecciones de heridas operatorias se han mantenido como la tercera infección más frecuente, donde los indicadores de infección de herida operatoria son en pacientes con operaciones específicas, donde se incluyen las operaciones de colocación de prótesis (MINSAL, 2019) . En diversos estudios se ha revisado el fenómeno de infecciones post operatorias en el sitio del implante. En un estudio de revisión que incluye el metaanálisis de 227 estudios con 1118 pacientes que experimentaron infecciones, el ritmo de infección total observado fue de 4, 87%, lo que fue más severo en los procedimientos neuroquirúrgicos. Se identificó la presencia de bacterias grampositivos, como S. aureus, Staphylococcus coagulasa-negativo y P. acnes (Chen, Y., et al., 2019) . En el documento de revisión de Kwarcinski J. y colaboradores se destaca una amplia variación en el riesgo de infección informado para diferentes materiales en la reparación craneal. Se señala que los materiales seleccionados buscan imitar el hueso natural y ayudar a restaurar la función (estructural y estética) del cráneo humano pero que no están ausentes del riesgo de producir una infección. De acuerdo con los resultados de la revisión, 41 artículos que cumplían con los criterios de inclusión) , las tasas de infección promedio por material oscilaron entre 2, 04% y 10, 98%. Los resultados indican que existe variación entre los materiales con respecto al riesgo total de infección, sin embargo, dependiendo de los materiales comparados, este valor puede ser insignificante. Los factores de riesgo alternativos asociados con la infección, incluido el tiempo quirúrgico, las revisiones y la infección previa, tienen un mayor impacto en el potencial de infección que la variación material. La comparación de los métodos de fabricación destacó un efecto notable en la tasa de infección promedio. Se pueden observar tendencias que muestran que los materiales con mayores niveles de interacción superficial y soporte activo del crecimiento del tejido presentaron mayor resistencia a la infección. En el caso del material PEEK, las tasas de infección variaron entre 0, 00% y 14, 29%, con una tasa de infección promedio de 7, 89%, una desviación estándar de 5, 16% y un riesgo relativo de 0, 75 (Kwarcinski et a/., 2017) . Otra de las dificultades observada es la inflamación de la zona operatoria por la presencia del implante. Las prótesis se han ido actualizando de tal manera que se disminuyen los riesgos asociados a infecciones, por ejemplo, el cemento óseo se ha ido mejorando de manera tal que posea propiedades mecánicas y antibacterianas por lo que se disminuye el riesgo de infección. No así para las prótesis no cementadas, las cuales en la actualidad poseen características limitadas para prevenir o tratar infecciones bacterianas, donde básicamente la estrategia a realizar es cirugía de revisión (Chengzhe et a/, 2021) . El uso de materiales como PEEK son una alternativa como material para la fabricación de implantes en aplicaciones clínicas. Este material es biocompatible, químicamente estable radiotransparente y tiene un módulo elástico similar al del hueso natural (Gu et al., 2021) . Se han descrito estudios en donde se evalúa el uso de prótesis fabricadas con PEEK que pueden tener efectos antimicrobianos luego de ser colocados en el paciente para poder disminuir los riesgos de infección post operatorio. Por ejemplo, Delaney y colaboradores en 2019 describieron un depósito fabricado con PEEK que podía ser incorporado a un paciente como prótesis de columna. Esta prótesis podía liberar de forma prolongada antibióticos y/u otros compuestos, donde dicho depósito posee poros y al momento de aplicar ultrasonido los compuestos contenidos en el depósito son liberados de forma controlada y prolongada (Hernández et al., 2020) . Se han realizado estudios sobre prótesis generadas con PEEK con adiciones de nanopartículas o aleaciones con otros materiales que podrían tener actividad antimicrobiana. El documento de Yan y colaboradores en 2018 describe la elaboración de un recubrimiento de nanopartículas de plata y sulfato de gentamicina sobre una superficie porosa de PEEK (Yan et al., 2018) . El recubrimiento mejoró la capacidad bactericida del PEEK sobre bacterias Gram positivo y Gram negativo, además el número de bacterias adheridas a PEEK con el recubrimiento fue menor que en las condiciones de PEEK sin recubrimiento (Yan et al., 2018) . Xue y colaboradores en 2020 desarrollaron un método de deposición capa por capa (LBL) con ciclos controlados para construir rápidamente capas de brushita (CaHPO42H2O) (CaP) que contienen sulfato de gentamicina (GS) en PEEK para obtener PEEK modificado con CaP y GS (PEEK/CaP-GS) (Xue et al., 2020) . Los autores describen que realizaron ensayos antimicrobianos in vitro observando que las muestras de PEEK/CaP-GS tenían una propiedad antibacteriana excelente y sostenida, mientras que en ensayos de proliferación celular demostraron una compatibilidad aceptable y diferenciación osteogénica celular (Xue et al., 2020) . Por otro lado, el documento de Lazar y colaboradores en 2016, desarrolló un implante reforzado con fibra recubierto con gentamicina para reconstrucción craneofacial con propiedades antimicrobianas. Los resultados de este estudio indicaron que las bacterias fueron inactivadas al entrar en contacto directo con los recubrimientos de gentamicina (Lazar et al., 2016) . Chengzhe y colaboradores, en su documento publicado en 2021 presenta un estudio en el cual se desarrolló un implante PEEK que tiene por función combatir simultáneamente la contaminación bacteriana y promover la osteointegración mediante la liberación sostenida de clorhidrato de moxifloxacino (MOX) y péptido de crecimiento osteogénico (OGP) a partir de poli dopamina con superficie recubierta de PEEK sulfonado poroso (SPK) (Chengzhe et al, 2021) . Los resultados indican que la superficie SPK (SPD-MOX/OGP) modificada con MOX/OGP-PDA exhibió un efecto antibacteriano duradero y excelente contra Staphylococcus aureus planctónico/adherente y Escherichia coli in vitro. Además, se observó una mejora notable en la adhesión celular específica, la proliferación y la osteogenicidad en el sustrato SPD-MOX/OGP debido a la presencia de moléculas OGP y PDA en comparación con todos los demás grupos (Chengzhe et al, 2021) . En documentos de patente también se han descrito implantes para el control bacteriano en el sitio del implante. Por ejemplo, el documento CN101432030B describe un cuerpo o depósito tridimensional que incluye una o más sustancias implantables. Dicho deposito puede recibir cantidades de un líquido. El depósito más el líquido pueden formar una masilla que puede ser útil como material implantable. Otro documento como WO2007001624A2 divulga un dispositivo médico implantable para el tratamiento de la osteonecrosis. Dicho dispositivo de implante adaptado para su inserción en uno o más canales o huecos en el tejido óseo; una pluralidad de depósitos discretos ubicados en la superficie del al menos un cuerpo del dispositivo de implante; y al menos un sistema de liberación dispuesto en uno o más de la pluralidad de depósitos, en el que el sistema de liberación incluye al menos un fármaco seleccionado del grupo que consiste en promotores del crecimiento óseo, promotores de la angiogénesis, analgésicos, anestésicos, antibióticos y combinaciones de los mismos. En el documento US20170056565A1 se divulga un clip biocompatible fabricado con PEEK para el tratamiento de infecciones sitio quirúrgicos, el cual va adherido a un implante. Este clip comprende un depósito y una abertura del depósito, donde esta última está sellada con un material sensible a la presión que puede romperse al aplicar una fuerza a dicho material y liberar de forma prolongada compuestos. El depósito puede ser llenado con un agente terapéutico seleccionado del grupo que consiste en: un antibiótico, antivírico, analgésico, factores de crecimiento, antifúngico, antimicobacterias, quimioterapéutico, osteogénico, como ascorbato para aumentar la ormación de hueso, hormona paratiroidea, péptidos, peptoides, AINE, analgésicos o combinaciones de los mismos. El documento US8821912B2 describe métodos para fabricar dispositivos médicos implantables, que pueden ser fabricados con PEEK y poseen propiedades antimicrobianas. El efecto antimicrobiano de estos dispositivos se produce por la incorporación de partículas cerámicas con cationes metálicos antimicrobianos en resina PEEK fundida, que posteriormente se deja enfriar y fraguar en su forma final lograda mediante moldeo por inyección, corte y mecanizado u otras técnicas. Si bien, en el estado del arte se han descrito y realizado avances en la fabricación de prótesis con propiedades antimicrobianas, aun se requieren alternativas mejores para evitar la generación de infecciones en el sitio del implante de forma prolongada y controlada. Es reconocido en la práctica clínica que las primeras 48 a 72 horas de la cirugía de incorporación del implante son vitales por lo que se requieren alternativas de implante que sean capaces de mantener un ambiente antimicrobiano prolongado durante este tiempo para evitar la generación de infecciones. Las propuestas existentes hasta ahora corresponden a implantes con acción antimicrobiana de contacto y momentánea, o que requieren la funcionalización con nanopartículas, lo que complica técnicamente y encarece el proceso de producción del implante. En otros casos, se acoplan clips o dispositivos externos en la superficie del implante, esto conlleva la complicación del proceso de producción y el ajuste espacial del implante complejo en el cráneo del paciente. Por lo tanto, existe la necesidad de un implante craneofacial que pueda liberar de forma constante y prolongada sustancias activas de interés e importancia clínica en la zona del implante. Esto permitiría en ciertos casos, ejercer o promover un ambiente antimicrobiano prolongado y controlado durante las primeras horas de fijación del implante para evitar la generación de infecciones, y que, a su vez, sea un implante simple y de menor costo de producir y de trabajar. En la presente solicitud se describe un implante, particularmente un implante craneofacial fabricado en PEEK, el cual comprende o incluye uno o más depósitos o ecámaras de almacenamiento que en su interior pueden almacenar principios activos o sustancias en estado líquido o formato líquido que son liberados de forma controlada y gradual por gravedad en el sector del reemplazo óseo. DESCRIPCIÓN DE LA INVENCIÓN La invención corresponde a un implante craneofacial de material Poliéter éter cetona (PEEK) para la liberación de principios o sustancias activas, porque dicho implante comprende: Uno o más depósitos o recámaras o cápsula interiores (1) ovaladas con paredes de 2, 5 a 4 mm de grosor, que almacenan un líquido o sustancia activa en formato líquido para ser liberado de forma gradual y prolongada al sitio del implante, tuberías (2) que interconectan los depósitos o recámaras interiores con los orificios de entrada y salida, definiéndose: a) una tubería de entrada que conecta el orifico de entrada (3) con el depósito (1) de tal forma que incluye un sistema de embudo que incluye una entrada o carga del líquido que tiene un diámetro de 1, 5 a 2, 0 mm y que luego se ensancha en una tubería de diámetro 2, 0 a 3, 0 mm para continuar con la tubería hasta el depósito. b) una tubería de salida que conecta el depósito o recámara (1) con el orificio de salida (4) que incluye un sistema de embudo de transición que presenta un diámetro de 0, 7 a 2, 0 mm en la parte superior y un diámetro de 0, 7 a 1, 5 mm en su parte inferior, dependiendo del diámetro del orificio de salida (4) , c) Uno o más orificios de entrada (3) en la parte superior del implante dependiendo del número de depósitos, donde dicho orificio es la entrada o carga del líquido para llegar a los depósitos (1) , donde el orificio de entrada tiene un diámetro de 1, 5 a 2, 0 mm y está bloqueado por un tapón de filamento PEEK o un tornillo de titanio, Uno o más orificios de salida (4) en la parte inferior del implante dependiendo del número de depósitos como orificio de salida gradual y prolongada del líquido o sustancia activa hacia el sitio del implante, donde el orificio de salida tiene un diámetro de 0, 7 a 1, 5mm y tiene un tapón o sistema que sella el implante para no permitir que el líquido se escape cuando no se ha dispuesto en el sitio del implante. El implante craneofacial de la presente invención evita las infecciones post-implante, particularmente, las infecciones bacterianas. Este efecto lo logra gracias a la liberación sostenida, controlada y prolongada de un líquido, sustancia o principio activos en formato líquido. Los inventores han definido específicamente el diseño y ubicación espacial de cada componente dentro del implante de tal manera de disponer de depósitos o recámaras lo suficientemente grandes para acumular una cantidad considerable de un líquido o principio activo o sustancia activa en esta forma, pero al mismo tiempo, que el implante no pierda sus características y propiedades estructurales. Los inventores llevaron a cabo una serie de iteraciones experimentales para definir las mejores condiciones estructurales de tamaño, longitud y diámetro de cada parte o componente del implante craneofacial de la presente invención. Fue un desafío técnico y de diseño el generar un implante con uno o más depósitos o sistemas de recámaras, más bien multicámaras de almacenaje interno para líquido con una capacidad apropiada. Más complejo e importante, cómo se añade y libera el líquido de interés desde el implante al paciente. Así, en las iteraciones se definió empíricamente que el depósito puede contener uno o más depósitos o sistemas de recámaras de almacenamiento dependiendo de la necesidad de vaciado del líquido a almacenar. Particularmente, se definieron implantes con un depósito de capacidad de 5, 0 mL de llenado e implantes con 2 o más depósitos para incluir al menos 2, 5 mL del principio activo o sustancia en formato o estado líquido. Aun cuando inicialmente se observó que era difícil manejar el flujo del líquido y las proporciones de los orificios de entrada y salida para lograr la salida controlada y prolongada en los implantes con múltiples o más cámaras, los inventores incluyeron en sus iteraciones experimentales la definición de cada diámetro de entrada y salida, en onjunto con las longitudes y disposición física de los componentes de cierre (tapón) que impiden la salida del líquido cuando aun no se ha utilizado el implante y permiten su correcto funcionamiento. Cuando los inventores evaluaron los diámetros de los orificios de entrada y salida, también se encontraron con diferentes problemáticas para lograr la liberación correcta y prolongada del líquido o principio activo depositado en las recámaras. En la presente invención las condiciones de los tamaños de depósito o cápsula y de orificios de entrada y salida guardan relación con la regulación de la salida o liberación del líquido o sustancia activa en formato líquido desde los depósitos o cápsulas al medio. Como parte de las iteraciones experimentales, los inventores definieron que los orificios de entrada en la parte superior del implante para la entrada o carga del líquido o sustancia activa, deben tener un diámetro apropiado para que se pueda acoplar una jeringa que introduzca dicho principio activo o líquido al implante. A su vez, el orificio no debe colapsar ni permitir la salida o sobrepaso del líquido. En ese sentido, los inventores han definido que la tubería de entrada que conecta el orifico de entrada (3) con el depósito (1) incluye un sistema de embudo que incluye una entrada o carga del líquido que tiene un diámetro de 1, 5-2, 0 mm y que luego se ensancha en una tubería de diámetro 2, 0 a 3, 0 mm para continuar con la tubería hasta el depósito, donde el orificio de entrada tiene un diámetro de 1, 5-2, 0 mm. Como parte de las mejoras se adicionó también una sección adicional en el orificio de entrada del implante para insertar un trozo de filamento y que funcione como tapón. Este tapón puede ser de material PEEK o puede corresponder a un tornillo de titanio. En el caso de los orificios de salida en la parte inferior del implante como orificio de salida gradual y prolongada del líquido o sustancia activa hacia el sitio del implante, los inventores observaron en las pruebas expuestas en los ejemplos de aplicación la necesidad de regular la salida del líquido desde los depósitos o cápsulas de tal forma de aumentar el tiempo efectivo de liberación y exposición el líquido en el ambiente o sector del implante. Es importante destacar que los inventores han definido todas las condiciones de sistema tipo embudo, diámetros de entrada y salida y las distancias y grosores mínimos de cada estructura y disposición de éstas en el diseño del implante, de tal forma de permitir la liberación prolongada de la sustancia activa en formato líquido. Así, los inventores definieron que el implante debe disponer de uno o más orificios de salida (4) en la parte inferior del implante dependiendo del número de depósitos como orificio de salida gradual y prolongada del líquido o sustancia activa en formato líquido hacia el sitio del implante, donde el orificio de salida tiene un diámetro de 0, 7 a 1, 5mm y tiene un tapón o sistema que sella el implante para no permitir que el líquido se escape cuando no se ha dispuesto en el sitio del implante. La regulación del tamaño del diámetro del orificio de salida y los tamaños definidos por los inventores para cada estructura del implante permiten la liberación gradual del líquido o sustancia activa den formato líquido. En el caso de las tuberías que se interconectan a los depósitos o recámaras, se disponen de tuberías en la parte superior de los depósitos o recamaras que originan los orificios de entrada para la incorporación del líquido a dichos depósitos y tuberías que van desde los depósitos a la zona inferior o parte de abajo del implante y que dan origen a los orificios de salida con diámetros controlados y definidos según lo ya descrito. Las tuberías de todo el sistema tanto de entrada como de salida deben de ser de al menos 10 mm, con un máximo de 15 mm, este mínimo se debe a las pruebas desarrolladas para que el líquido pueda entrar de manera adecuada a la recámara o depósito. En una de las formas de la invención, la liberación del líquido o sustancia activa en formato líquido se relaciona directamente con la entrega de sustancias activas en el sitio del implante, luego de su inclusión en el cuerpo del paciente. En ese sentido, la liberación de sustancias activas en formato líquido desde el implante al sitio de implante permite generar un efecto específico y prolongado en el lugar del implante luego de la cirugía. En una de las formas de la invención, la liberación prolongada y sostenida durante los primeros días luego del implante permite evitar y disminuir infecciones del tipo postoperatorias. La liberación prolongada de sustancias activas las primeras horas postoperación, como sustancias antibióticas y antiinflamatorias, evita la colonización de bacterias que pueden causar alguna infección en el sitio del implante. Tal como se demuestra en los ejemplos de aplicación, en particular en la figura 1 parte del ejemplo de aplicación 1, el implante libera por goteo. Los inventores definieron los tamaños específicos para la liberación por goteo como un mecanismo de liberación prolongada en la zona del implante. En una de las formas de la invención, la liberación de sustancias o principios activos en formato líquido desde el implante permite un efecto clínico local dependiendo del tipo de principio activo, sin limitarse, a antibióticos, antiinflamatorios, analgésicos u otro tipo de principio activo. Se incluyen también como principio activo agentes quimioterapéuticos o cualquier principio activo soluble que pueda conformar una solución. Como parte del alcance de la invención, se puede incluir al menos un principio activo, siendo posible poder incluir más de uno, donde estos se pueden incluir en el mismo depósito o en depósitos separados. Los inventores han demostrado en los ejemplos de aplicación 2 y 3 que los implantes parte del alcance de la invención pueden liberar de forma prolongada un líquido o sustancia activa en formato líquido desde uno o más depósitos o recámaras o cápsulas incluidas en la estructura interna del implante. La disposición física, espacial y los tamaños de cada componente del implante permiten la liberación de la sustancia líquida desde las 24h, hasta incluso 11 días luego de incluir en él algún tipo de sustancia en este formato. Incluso, en condiciones simuladas de movimiento (simulación del movimiento de la cabeza del paciente) se apreció la liberación sostenida y prolongada de la sustancia líquida desde el implante al medio, por varios días. Estos resultados demuestran que el implante parte de la invención es funcional y libera de forma sostenida y prolongada el líquido o sustancia en formato líquido incluida en él o los depósitos del implante. Es importante destacar que los resultados expuestos en los ejemplos de aplicación 1 a 6, en particular para los ejemplos 5 y 6, si bien fueron evaluados para vancomicina como principio activo, la prueba y resultados es extrapolable y válida para cualquier otro principio activo que se pueda solubilizar en algún solvente apropiado, en este caso solventes o vehículos farmacéuticamente aceptables. Un ejemplo de solvente puede ser agua, suero fisiológico, buffer fosfato dibásico de potasio, buffer fosfato salino o cualquier otro buffer o vehículo aceptable para su administración a un ser vivo, en particular a un ser humano. En ese sentido, es parte del alcance de la invención la posibilidad de incluir cualquier tipo de principio activo que sea soluble en un vehículo farmacéuticamente aceptable según lo ya señalado. En el ejemplo 5 se expone el análisis de la concentración y tasa de liberación. La cantidad de vancomicina liberada en la primera hora fue de 13.087, 5 g. Luego en la hora 2 se observa una diminución de la cantidad liberada con 548, 5 g pero que luego se retoma o aumenta en la hora 4 con 1.232, 5 g del principio activo. Para las horas 8 y 12 del ensayo se observó también un aumento con 3.505, 5 g y 4.309, 4 g, respectivamente. Respecto de la tasa de liberación, en la primera hora se observa una alta tasa de liberación de vancomicina con un promedio de 218 g/mL, la que luego disminuye en el tramo de tiempo entre la primera y segunda hora, para luego aumentar y mantener la liberación sostenida. Esta cinética de liberación permite indicar que primero hay una liberación rápida o mayor del principio activo para luego mantener una liberación paulatina y constante. En el ejemplo de aplicación 6 se demuestra que el principio activo cargado en el o los depósitos del implante se mantiene estable y no pierde su estructura ni se degrada, por lo que es funcional. En el caso particular, se evaluó la estabilidad de una solución de vancomicina en el implante por 24 horas, observándose que no existe degradación significativa del principio activo por lo que el depósito no afecta a la estabilidad del principio activo a liberar. Esta información permite señalar que el principio activo se libera de forma correcta y con una cinética de liberación apropiada para la liberación de principios activos o fármacos en la zona del implante craneofacial, donde se observa primero la salida rápida para lograr el efecto terapéutico, una disminución y luego la mantención de la liberación por al menos las primeras 43-48 horas. Además, la puesta del principio activo o solución de éste en el depósito del implante no afecta su estabilidad. El implante descrito en la presente invención corresponde a un implante craneofacial fabricado mediante impresión 3D en material PEEK customizado. Dicho implante comprende dos orificios o aberturas, una superior y una inferior que permiten el llenado y salida de las sustancias a liberar, respectivamente, tal como se ha descrito para la presente invención. En las realizaciones de la invención, los receptáculos o depósitos o cámaras o reservorios descritos en el implante, pueden incorporar sustancias o medicamentos o principios activos en formato o estado líquido, los cuales pueden ser liberados en el sector donde se realizó el reemplazo óseo. En realizaciones de la invención, los implantes con depósitos o recámaras o reservorios incorporan particularmente medicamentos o principios como antibióticos y/o antiinflamatorios y/o analgésico con el fin de llevar al éxito la incorporación de dicho implante. Es también parte del alcance de la invención que el medicamento o principio activo corresponda a un agente quimioterapéutico o cualquier otra sustancia que se pueda disolver y obtener como una solución líquida. Es importante también señalar que debido a que el implante puede tener más de un depósito, también es parte del alance de la invención La liberación de sustancias a través del receptáculo, depósito cámara o reservorio del implante customizado con PEEK, descrito en la presente invención se debe a la acción de la gravedad, la cual permite la liberación gradual de dichos compuestos. La liberación depende del diámetro del orificio de salida y demás tamaños de las estructuras del implante. Dentro de las realizaciones de la invención, el desarrollo del implante customizado incluye depósitos o recamaras interiores y con un sistema interconectado de tuberías, donde dichas recamaras pueden almacenar sustancias liquidas las cuales pueden ir siendo liberadas por la fuerza de gravedad. Particularmente, se presenta un implante con depósitos o recamaras superiores e inferiores que permiten el paso por gravedad de las sustancias, donde dichas sustancias pueden ser antibióticos y/o antiinflamatorios y/o analgésicos. En realizaciones de la invención, se describe que las sustancias liquidas que pueden ser incorporadas en las recamaras internas del implante mediante el uso de una jeringa quirúrgica. Definiciones El presente documento describe un implante craneofacial, donde este último se refiere a cualquier implante que puede ser colocado en un paciente en las zonas del cráneo y/o cara. El implante craneofacial de la presente invención permite la liberación prolongada y sostenida de principios activos o sustancias activas, particularmente en forma líquido, para generar un efecto específico terapéutico en la zona del implante. Se incluyen diferentes tipos de sustancias o principios activos en busca de efectos de analgesia, antinflamatorio u antibiótico, agentes quimioterapéuticos o cualquier principio activo capaz de incluirse en una solución. En una de las formas de la invención, permite evitar las infecciones post-implante, particularmente, las infecciones bacterianas. Una medida de esto es la diminución del recuento bacteriano. En ese sentido, cuando se ha referencia a la disminución del recuento bacteriano, se hace referencia a la disminución en el recuento de UFC o UFC/mL de las bacterias. Cuando en el documento se hace referencia a PEEK customizado, se indica que el implante descrito en la presente invención es fabricado en PEEK y es personalizado según las características del paciente que recibirá el implante. Cuando en el documento se indica el término "receptáculo", "cámara", "deposito", "reservorio" y/o "recamara interior" y/o "cápsula" se indica una cavidad en que se contiene o puede contener una sustancia en su interior. El término "postoperatorio" se refiere al periodo que sigue luego de una intervención quirúrgica y que finaliza con la rehabilitación del paciente. El término "perioperatorio" corresponde al periodo de tiempo de un procedimiento quirúrgico, el cual comprende desde la preparación del paciente hasta su recuperación. Cuando en el documento se indica "liquido activo", "principio activo" o "sustancia activa" se hace referencia a toda sustancia o mezcla de estas que poseen una función y/o actividad particular en la composición de un medicamento. En el documento se indica el término "iteraciones", este término hace referencia a una repetición de pasos para un determinado fin. En el caso de la presente invención, las iteraciones corresponden a la repetición de la fabricación del implante y las realizaciones e pruebas sobre este para llegar a obtener el implante con la funcionalidad que se requiere. Descripción Figuras Figura 1.- Imagen prototipo iteración 1 para desarrollo de implante. Se presenta el prototipo 1 compuesto de múltiples cámaras de depósito. Fallo de goteo y salida del líquido desde el reservorio. A) presenta la estructura del prototipo de implante; b) se describen las partes del prototipo, donde (1) recamaras de contención del líquido o principio activo con capacidad total de 3 mL, (3) entrada del líquido o principio activo mediante jeringa con diámetros de 0, 8, 1, 25 y 1, 25 mm; y (4) orificios de salidas con diámetro de 0, 5, 0, 7 y 1 mm para el líquido o principio activo. Figura 2.- Imagen prototipo iteración 2 para desarrollo de implante. En A) Se presenta el Prototipo 2 de implante con una sola recámara o depósito (1) . B) de describe la entrada del líquido o principio activo por una tubería de 26 mm de largo (2) y el espesor del prototipo de 4, 7 mm se indica con una flecha negra gruesa. Prototipo 2 falló pues el implante pierde capacidades mecánicas, al debilitarse la estructura interna del implante. Figura 3.- Imagen prototipo de iteración 3 para desarrollo de implante. Implante prototipo 3 con 2 recámaras independientes para el depósito de líquido o principio activo. Prototipo fallido por problemas en el diámetro de salida de la tubería de uno de los depósitos. En (1) se indican los depósitos de líquido o principio activo; (3) orificios de entrada del líquido o principio activo por medio de una jeringa; (4) orificios de salida del líquido o principio activo. Figura 4.- Imagen prototipo de iteración 4 para desarrollo del implante. Se agrega material en la parte interna para tener al menos 2, 5 mm de grosor en cada lado de la recámara o depósito (1) . En (2) diámetro de tubería de 1, 4 mm, (3) el orificio de entrada para un diámetro de jeringa de 1, 5 mm, (4) orificio de salida de diámetro de salida de 0, 75 mm, o 0, 5 mm. Figura 5.- Imagen de iteración 5 para desarrollo del implante. Se provee un implante con 2 cámaras o depósitos internos con pares de separación de 3, 5 mm de grosor y con os orificios de entrada y salida de líquido definidas. A) esquema digital del implante, b) molde del implante. Figura 6.- Imagen de iteración 6 de mejora del implante: tapón en orificio tubería de ingreso del líquido al implante y sistema tipo embudo. En A) se presenta una imagen de la parte superior del implante donde se aprecia la tubería de entrada y el orificio de entrada donde se dispone un trozo o pieza de PEEK para funcionar como un tapón que impide que el líquido contenido en el interior del implante se escape o devuelva por el orificio de entrada. En B) se presenta un implante que incorpora mejoras como un sistema tipo embudo en el ingreso del líquido, este sistema va variando los diámetros de cada cañería según lo que se muestra en la imagen con 1, 5 mm para la entrada de la aguja de la jeringa que incorpora el líquido al implante y luego un ensanche en una tubería de 3, 0 mm en la tubería hacia el depósito o recámara. En dicho implante, el orificio de entrada expone una tubería de salida con un sistema embudo que tiene dimensiones de 2, 0 mm para luego disminuir su diámetro a 1, 3 mm en el orificio de salida. El implante de la imagen tiene un solo depósito de 5 mL de capacidad con paredes de un grosor de 2, 5 mm de espesor. En 6C) se expone el tapón o sistema que sella el implante en el orificio de salida para no permitir que el líquido se escape cuando no está en uso (ver figura 6C) . Figura 7.- Imagen de iteración 6 de mejora del implante: tapón en orificio tubería de ingreso del líquido al implante y sistema tipo embudo en salida. En A) Se expone el prototipo de implante con un depósito de medidas 52, 2 mm de ancho y 52, 86 mm de alto para el depósito. En B) se expone un prototipo de implante de dos depósitos o recámaras que presenta una recámara izquierda de 24, 43 mm de ancho por 46, 7 mm de alto y un recámara o depósito izquierdo con un ancho de 24, 47 mm y 46, 84 mm de alto. Figura 8.- Fotografía Ensayo de liberación de líquido desde un implante de la presente invención (ejemplo de aplicación 2) . Se observan las tonalidades diferentes de azul de acuerdo con la liberación del azul de metileno diluido en agua cuando el implante se dispone en un recipiente. Se valuaron de izquierda a derecha los tiempos de liberación de 1, 2, 3, 4 y6 horas. Figura 9.- Fotografía Prueba de verificación del tiempo de vaciado en implante de un depósito (ejemplo de aplicación 3) . En A) se expone una fotografía de los resultados de la observación de las muestras obtenidas en los puntos de tiempo de 24, 72, 96, 120 y 144 horas. De izquierda a derecha, en el tiempo de 24 horas se observó muy poca coloración del medio o salida del líquido desde el implante. En horas intermedias de 72, 96 y 120 horas se observa mayor liberación del líquido con una tonalidad celeste suave observada en el ambiente (recipiente) para que a las 144 horas la tonalidad cambie a una tonalidad celeste fuerte. Cuando se evaluó el tiempo de 168 horas se observa una mucho mayor liberación del contenido desde el reservorio o cápsula dentro del implante al medio (recipiente) . En B) se expone una fotografía del orificio de salida o liberación del líquido contenido en el implante, en este caso azul de metileno diluido en agua. Se observa la salida del líquido desde el orifico de salida del implante con una coloración azul más fuerte. Figura 10.- Imagen implante con depósitos y tuberías para la liberación gradual de un líquido o principio activo al cerebro. En A y en B se presenta la imagen del implante integrado en el cráneo del paciente. En A, se observa el detalle del interior del implante. En B, el implante es visto desde la parte interna del cráneo, en donde las salidas de las recamaras liberan hacia el interior del cerebro el líquido. Figura 11.- Gráfica vancomicina liberada (g) desde la prótesis de acuerdo con el tramo de tiempo evaluado. Se presenta la gráfica representativa de esta cinética de liberación considerando los g liberados para los puntos de muestreo de 0, 2, 4, 8 y 12 horas. Figura 12.- Tasa de liberación de vancomicina desde el implante por tiempo de muestreo. Se expone la gráfica de la liberación de g/min de acuerdo con el tiempo de muestreo evaluados de 0, 2, 4, 8 y 12 horas. Ejemplos de aplicación Ejemplo 1: Desarrollo de prototipos de implantes con depósitos y tuberías para liberación controlada de líquidos o sustancias activas Es parte del presente ejemplo de aplicación, el desarrollo del prototipo de implante considerando las iteraciones y análisis experimentales correspondientes para obtener n implante que conste de depósitos o recamaras interiores interconectados con tuberías de tal forma de poder incluirse en él, un líquido o sustancia activa, pero también que se libere de forma prolongada durante las primeras 24-48 horas luego de incorporar el implante al paciente. Para las pruebas se utilizó un implante 3D previamente diseñado, al cual se le adicionó uno o más depósitos o recámaras de almacenaje interno de líquido. - Primera iteración de prueba: Implante con más de un depósito o sistema de recámaras El primer prototipo de implante que se desarrolló contaba con varios depósitos o recamaras interiores y con un sistema interconectado de tuberías, con el fin que cada recamara almacenara líquido (prueba con antibiótico) y por gravedad se fuera trasladando este líquido desde las recamaras con mayor cantidad a las de menor cantidad. Este prototipo contaba con tres entradas de líquido a través de una cañería recta, con un diámetro de 0, 8 mm; 1, 25 mm y de 1, 5 mm. En ese sentido, los inventores observaron que el diámetro de entrada es de suma importancia pues debe ser lo suficientemente grande para permitir cargar correctamente el líquido al interior con una jeringa, pero al mismo tiempo, no ser tan grande para que en el proceso de carga el líquido no rebote y se salga del implante. De acuerdo con las pruebas de este prototipo, el mejor diámetro fue de 1, 5 mm. Este primer prototipo funcionó respecto de la salida o liberación de líquido pues se observaron varias salidas de líquido por goteo, pero al ser un sistema de cañerías tan extenso y con diámetros delgados ocurrió que algunas de estas salidas no funcionaron. Las salidas evaluadas fueron de diámetros 0, 5 mm; 0, 7 mm y 1 mm. En este caso aun cuando el implante no funcionó por completo de forma correcta, se observó que la salida de 0, 7 mm permite la salida controlada de goteo. La salida de 0, 5 mm se obstruía y la salida de 1 mm presentaba un flujo muy alto de salida (figura 1A y 1B) . En la tabla 1 se resumen los resultados de la definición de los diámetros de los orificios de entrada y salida del líquido a incluir en el implante para ser liberado al sector de inserción del implante. Tabla 1. Resultados determinación orificios de entrada y salida del implante. - Segunda iteración de prueba: implante con un depósito o recámara Para las pruebas se utilizó un implante 3D previamente diseñado, al cual se le debió adicionar un depósito o sistema de almacenaje interno de líquido. En primer lugar, se diseñó el sistema de cañerías y la forma de unión o interacción con el depósito o recámara. Se proveyó una entrada de líquido que se dirige hacia la recámara y con una salida que se encuentra sobre él, esta salida posee una forma piramidal para poder controlar la velocidad y cantidad de líquido que se deposita. Luego, otro de los desafíos de diseño fue definir la capacidad interna del depósito o recámara. La primera prueba se realizó con un prototipo de implante que incluyó un depósito de capacidad interna de 2 mL. Se produjo un implante de un tamaño de 85, 62 mm x 99, 7 mm con un espesor de 4, 7 mm, compuesto de una sola recámara o depósito con una tubería de 26 mm de largo conectada a él (ver figura 2A y 2B) . Para evaluar el funcionamiento del implante, en particular en la liberación de un líquido o principio activo, se realizó una prueba adicionando agua líquida al implante por medio de una jeringa. Se observó que el depósito único de 2 mL es muy grande pues el implante pierde capacidades mecánicas, al debilitar la estructura interna del implante. Para solucionar esta problemática y que el implante presente propiedades mecánicas adecuadas, se estableció en el diseño que las paredes de cada depósito deben tener al menos 2, 5 mm de grosor. Cuando se probaron implantes con estas consideraciones, los implantes mantenían sus propiedades mecánicas. - Tercera iteración de prueba: Mejora implante con más de un depósito o recámaras Se realizó un segundo prototipo o mejora de los implantes con dos depósitos o recamaras independientes, cada una de estos dispone de una entrada de líquido y una salida. Cada recámara tiene una capacidad de almacenamiento de al menos 2 mL, por lo que la suma de las capacidades de las recamaras puede llegar a hacer de alrededor de 5 mL. El implante utilizado es alrededor de 20% más grande que el anterior, pues se requiere una mayor superficie para tener mayor capacidad de almacenamiento de líquido y no disminuir las propiedades mecánicas del implante. El implante comprende una tubería conectada a cada depósito, donde dicha tubería tiene una entrada del diámetro de una jeringa de 1, 2 mm. El implante tiene además unas tuberías de salida de un diámetro de 0, 5 mm para un depósito y de 0, 35 mm para el otro depósito (figura 3) . La prueba en este caso fue parcialmente exitosa ya que solo una de las dos recamaras cumple el objetivo de dejar fluir el líquido en forma de goteo, la otra entrada al ser de un tamaño tan pequeño por el método de fabricación se encuentra totalmente tapada. - Cuarta iteración de prueba La tercera iteración se basa en el mismo prototipo de implante anterior, pero con una gran diferencia, se agrega material en la parte interna para tener al menos 2, 5 mm de grosor en cada lado de la recámara o depósito. Esto de acuerdo con lo observado en la egunda iteración de prueba, mejora las condiciones y propiedades mecánicas del implante. Como parte de este prototipo, se aumentó el diámetro de la tubería de salida del líquido, pero para poder lograr el efecto y que sea una liberación controlada, en la parte superior se incluyó un sistema de embudo (figura 4) . Este implante tiene dos diámetros de salida diferente, de 0, 75 mm y 0, 5 mm. Se observó que el mejor diámetro de salida es de 0, 75 mm. En el caso de los 0, 5 mm el líquido no fluye de forma simple por lo que es difícil controlar el flujo del líquido. Al realizar una incorporación de material en las zonas de las recamaras se logró que el implante no disminuya sus capacidades mecánicas, logrando obtener una excelente capacidad de almacenaje en ambas recámaras sin afectar la calidad e integridad del implante. - Quinta iteración de prueba En una iteración posterior del prototipo previo, se procedió a disponer las recamaras de almacenamiento en la parte interior del implante conservando al menos 3, 5 mm de grosor entre la recamara y la parte exterior del implante. Para obtener el mismo grosor en la parte interior del implante es que se realizó un proceso de engrosamiento de la recamara, así obtener al menos 3, 5 mm de grosor, esta prueba resulto un completo éxito (figura 5) . Por lo tanto, el implante puede comprender solo un depósito de almacenamiento el que comprende paredes de un grosor de 2, 5-4 mm., preferentemente de 3, 5 mm. - Sexta iteración de prueba: Definición final sistemas de entrada y salida del líquido Considerando todas las problemáticas observadas en las iteraciones previas respecto al control de entrada y salida del líquido a liberar en el lugar el implante, se realizaron diversas mejoras al implante prototipo. Tapón orificio en tubería de entrada del líquido a almacenar En la sección inicial de la tubería por donde entra el líquido se adicionó al implante un trozo o pieza de la misma materia prima con la que se desarrolla el implante (PEEK) para funcionar como un tapón que impide que el líquido contenido en el interior del implante se escape o devuelva por la entrada. El filamento utilizado tiene un diámetro de 1, 75 mm, pero los termoplásticos tienen una dilatación por ello la entrada es 0, 25 mm mayor que la del filamento. Por lo tanto, el diámetro adecuado es de 2, 0 mm para bloquear la salida con el filamento (ver figura 6A) . Cuando se instala el implante, este trozo o filamento puede ser eliminado fácilmente (remover, quitar, traccionar) y una vez dispuesto el implante en su sitio, esta apertura queda cubierta por el hueso de la zona del implante. En este caso también es posible técnicamente incluir un tornillo de titanio de 1.5 mm que se enrosca en la salida para ser también un tapón o sello. Sistema embudo en la tubería de ingreso del líquido Se incorporó un sistema tipo embudo en el ingreso del líquido, este sistema va variando los diámetros de cada cañería, tiene como finalidad lograr que el líquido en el interior no se devuelva. Es un segundo método para impedir que el líquido se escapa del sistema de tuberías por el orificio de entrada. El sistema embudo conlleva que la tubería tenga un diámetro de 1, 5 mm para la entrada de la aguja de la jeringa que incorpora el líquido al implante y luego se ensancha en una tubería de 3, 0 mm. Es decir, existe un punto de embudo o transición para que el líquido no se devuelva (ver figura 6B) . Sistema de salida en la tubería de salida del líquido hacia el sitio del implante Se mejoró el sistema de salida, definiéndose que el diámetro del orificio de salida del líquido puede ir desde los 0, 7 mm hasta 1, 5 mm dependiendo de la geometría del implante. En este nuevo prototipo la tubería final de salida del líquido incorpora un sistema tipo embudo que busca favorecer que el vaciado de la recamara sea aún más controlada y prolongada (ver figura 6B) . Además, en esta prueba se adicionó un tapón o sistema que sella el implante para no permitir que el líquido se escape (ver figura 6C) , una vez que el implante se encuentra en pabellón, el equipo quirúrgico debe cortar una parte del cilindro donde quedará expuesto el orificio de salida para que el líquido pueda fluir desde el implante al interior del cuerpo. Medidas generales implantes y depósitos En cuanto a los tamaños, el implante en su extensión total dependerá de la zona del implante y de cada paciente. En el caso del tamaño del depósito o recámaras se definieron y evaluaron algunas dimensiones y tamaños generales. En el prototipo de implante de un depósito (a utilizar en ejemplo de aplicación 3) las medidas de 52, 2 mm de ancho y 52, 86 mm de alto para el depósito (figura 7A) . En el prototipo de implante de dos depósitos o recámaras evaluado presentaba una recámara izquierda de 24, 43 mm de ancho por 46, 7 mm de alto y un recámara o depósito izquierdo con un ancho de 24, 47 mm y 46, 84 mm de alto (figura 7B) . Las tuberías de todo el sistema tanto de entrada como de salida deben de ser de al menos 10 mm, con un máximo de 15 mm, este mínimo se debe a las pruebas desarrolladas para que el líquido pueda entrar de manera adecuada a la recámara de depósito. Ejemplo 2: Ensayo de liberación de líquido desde un implante de la presente invención Con el objetivo de estimar el tiempo de vaciado de la recámara o depósito se ha realizado un implante por impresión 3D, el que se compone de un sello de filamento de entrada de 2, 0 mm para bloquear la salid del líquido, una tubería de entrada con un sistema de embudo con un diámetro de entrada de 1, 5 mm para el ingreso de una jeringa y un diámetro posterior o de salida de 3, 0 mm. El implante probado tiene una cámara o depósito de capacidad de 5 mL con paredes de 2, 5 mm de espesor o grosor. La tubería de salida presenta un orificio de salida con un sistema embudo o de liberación controlada con una tubería de diámetro de 2, 0 mm con una transición a un diámetro de salida de 1, 3 mm (figura 6 b) . En la experiencia, el implante se llenó con 5, 1 mL de líquido en su interior, donde dicho líquido de prueba fue una solución de 4 partes de agua con una parte de azul de metileno. El protocolo consistió en inyectar los 5, 1 mL de la solución en la entrada del implante ya definido y sumergir el implante en un recipiente con agua, conservando la disposición geométrica de como seria instalado el implante. Luego, el implante se dejó en ese recipiente por un tiempo indeterminado, corroborando cada 1 hora si el color del agua ha cambiado con el colorante. Para las condiciones de prueba, se realizó la experiencia en un estado estático, es decir, sin movimiento del implante, o de forma dinámica simulando el movimiento de la cabeza del paciente. Esto último permite revisar si esto influye en la velocidad de la salida del líquido desde el interior del implante. Se observó que en la primera hora el líquido colectado desde el recipiente era casi transparente con una tonalidad o coloración celeste muy leve. A las 2 y 3 horas se observa el aumento de la tonalidad celeste como una tonalidad celeste suave. Luego de 4 horas se observa una tonalidad celeste fuerte indicando la salida del líquido introducido en el implante. Finalmente, con las pruebas realizadas hasta este momento el tiempo de vaciado sin movimiento es de aproximadamente 6 horas (ver figura 8 y tabla 2) . Cabe destacar que transcurrido este tiempo o más llegando a un límite de una semana, si el sistema se mueve el implante aún sigue liberando solución. Tabla 2.- Resultados observados experiencia liberación prolongada de líquido desde implante. Ejemplo 3: Prueba de verificación del tiempo de vaciado en implante de un depósito Se realizaron múltiples pruebas para verificar el tiempo de vaciado de la cápsula de 5 mL que posee el implante de prueba de una recámara. Se realizan además ensayos en un modo estático y en movimiento. Para los análisis se estandarizó y preparó un implante con las siguientes características técnica: • Diámetro de tapón de 2, 2 mm • Diámetro de inserción 1 mm (diámetro para insertar aguja) • Sistema embudo para disminuir la salida de líquido por la entrada en el implante • Capsula de 5mL • Paredes de al menos 2, 5 mm de grosor en todas las zonas de la cápsula o depósito • Salida de 1, 4 mm • Salida en forma de embudo para aumentar el tiempo de vaciado de cápsula o depósito Ensayo estático El experimento en su primera etapa consistió en llenar la cápsula o depósito del implante con una solución de azul de metileno diluido con agua en relación 5:1, este líquido es insertado dentro del implante verificando que la totalidad de la capacidad de la recámara o cápsula sea llenada. Luego en un recipiente plástico se procede a llenar con agua, se inserta el implante con el líquido en su interior y se ancla a un par de enganches metálicos dispuestos en el recipiente plástico para mantener una posición vertical, asemejando como este dispositivo estará posicionado en la realidad. Se evaluaron los siguientes tiempos: 24h posterior al inicio del experimento (primera muestra) , 72h posterior al inicio del experimento (segunda muestra) , 96h posterior al inicio del experimento (tercera muestra) , 120h posterior al inicio del experimento (cuarta muestra) , 144h posterior al inicio del experimento (quinta muestra) , 168 horas posterior al inicio del experimento (sexta muestra) . Los resultados de la observación de las muestras obtenidas en cada punto de tiempo permiten indicar que existe a simple vista un evidente aumento de la concentración del líquido liberado de forma prolongada al ambiente, en este caso al recipiente (figura 9A y 9B) . En la tabla 3 se describen los resultados de observación de las muestras obtenidas en cada punto de tiempo. En el tiempo de 24 horas se observó muy poca coloración del medio o salida del líquido desde el implante. En horas intermedias de 72, 96 y 120 horas se observa mayor liberación del líquido con una tonalidad celeste suave observada en el ambiente (recipiente) para que a las 144 horas la tonalidad cambie a una tonalidad celeste fuerte. Cuando se evaluó el tiempo de 168 horas se observa una mucho mayor liberación del contenido desde el reservorio o cápsula dentro del implante al medio (recipiente) . Cabe destacar que al día 11 aún sigue existiendo un goteo débil en la salida del implante, que sigue cambiando la concentración de azul de metileno en el agua del recipiente plástico. Tabla 3.- Resultados prueba de verificación del tiempo de vaciado en implante de un depósito o cápsula. Ensayo dinámico (implante en movimiento) Luego de realizar la prueba en un medio estático, se implementó una nueva prueba cuyo objetivo es recrear de manera más precisa cómo este dispositivo médico de tipo implante se comportará cuando sea usado por un paciente. En ese sentido, se recrearon los movimientos propios que ocurren en una persona (girar la cabeza, levantarse, caminar) . Para esto se utilizó una maquina 3D que tiene la capacidad de desplazar su plataforma en sentido horizontal. Sobre dicha máquina se posicionó el recipiente que contiene el agua y el implante relleno con la solución de azul de metileno anteriormente descrita en el ejemplo 2. El tipo de implante utilizado en la prueba también corresponde a un implante con las características descritas en el ejemplo de aplicación 2. Características del experimento: • Distancia de desplazamiento: 23 centímetros • Velocidad de desplazamiento: 3, 3 (cm/s) o 0, 1188 (km/h) • Ciclo corresponde al movimiento desde la posición inicial pasando por la posición final (23 centímetros) volviendo a la posición inicial, distancia recorrida 46 centímetros. • Cantidad de ciclos por minuto: 3. • Periodo de funcionamiento: 48 h. Luego de transcurridas 48h se detuvo la prueba. Transcurrido este tiempo, se observó que la concentración de azul de metileno en el medio a las 48 horas es mayor que lo observado para el tiempo 72 h en la experiencia sin movimiento (ejemplo 2) . Ejemplo 4: Implante craneofacial para evitar infecciones post-implante Luego de las iteraciones planteadas en el ejemplo de aplicación 1, se determinaron las mejores condiciones, tamaños y dimensiones del implante para poder incluir y liberar de forma gradual y correcta un líquido de interés (antibiótico y/o analgésico) . Se provee un implante craneofacial de material Poliéter éter cetona (PEEK) para evitar y disminuir las infecciones postoperación en el sitio del implante, porque dicho implante comprende: Uno o más depósitos o recámaras interiores (1) ovaladas con paredes de 2, 5 a 4 mm de grosor, que almacenan un líquido o sustancia activa en formato líquido para ser liberado de forma gradual y prolongada al sitio del implante, tuberías (2) que interconectan los depósitos o recámaras interiores con los orificios de entrada y salida, definiéndose: a) una tubería de entrada que conecta el orifico de entrada (3) con el depósito (1) de tal forma que incluye un sistema de embudo que incluye una entrada o carga del líquido que tiene un diámetro de 1, 5-2, 0 mm y que luego se ensancha en una tubería de diámetro 2, 0 a 3, 0 mm para continuar con la tubería hasta el depósito. b) una tubería de salida que conecta el depósito o recámara (1) con el orificio de salida (4) que incluye un sistema de embudo de transición que presenta un diámetro de 0, 7 a 2 mm en la parte superior y un diámetro de 0, 7 a 1, 5 mm en su parte inferior, dependiendo del diámetro del orificio de salida (4) , Uno o más orificios de entrada (3) en la parte superior del implante dependiendo del número de depósitos, donde dicho orificio es la entrada o carga del líquido para llegar a los depósitos (1) , donde el orificio de entrada tiene un diámetro de 1, 5-2, 0 mm y está bloqueado por un tapón de filamento PEEK o un tornillo de titanio, Uno o más orificios de salida (4) en la parte inferior del implante dependiendo del número de depósitos como orificio de salida gradual y prolongada del líquido o sustancia activa hacia el sitio del implante, donde el orificio de salida tiene un diámetro de 0, 7 a 1, 5 mm y tiene un tapón o sistema que sella el implante para no permitir que el líquido se escape cuando no se ha dispuesto en el sitio del implante. El implante integrado en el cráneo del paciente se acopla liberando de forma prolongada y continua el líquido contenido en los depósitos o recámaras, gracias al efecto de la gravedad y diseño del implante. Las salidas de las recámaras liberan hacia el interior del cerebro el líquido (figura 10A y 10B) . Ejemplo 5: Estudio analítico de la liberación de vancomicina esde el implante craneofacial En este estudio se revisó y evaluó la liberación de una solución de vancomicina desde el reservorio de un implante craneofacial parte del alcance de la invención. Este ensayo fue realizado en una incubadora que se encontraba a 37 grados centígrados y que realizaba un movimiento de 20 rpm, esto para imitar el movimiento que tiene un paciente luego de este tipo de cirugía. Para detectar la liberación y concentración de la solución de vancomicina liberada desde el implante, se utilizó Cromatografía Líquida de Alta Eficiencia (HPLC) acoplado a detector PDA. Como estándar se utilizó vancomicina clorhidrato con una concentración nominal de 0, 01 mg/mL preparado en suero fisiológico. Se generó una curva de calibración y la determinación de los tiempos de retención y valores de área de cada pico que aparezca en el cromatograma para determinar la concentración de vancomicina v/s el estándar. Para los muestreos como tal de la solución liberada desde el implante, se tomó una muestra de 0, 5 mL y se diluyeron en 0, 1 mL de suero fisiológico para luego llevar a un matraz de 100 mL con buffer fosfato dibásico de potasio 0, 2M a pH 7, 4. Esto se inyecta en el equipo para su análisis. Con los tiempos de retención y la determinación del área se determinó la concentración de vancomicina en la alícuota de análisis y se calculó la cantidad de vancomicina total presente en el vaso de forma acumulada (g en vaso) . Se realizaron mediciones a las 1, 2, 4, 8 horas, los resultados de estas mediciones se exponen en la tabla 4. Tabla 4.- Área detectada y cantidad de vancomicina de acuerdo tiempo de muestreo. Con este muestreo y calibración inicial se realizó un análisis de la cantidad de vancomicina liberada por tramos de tiempo. En la tabla 5 se presenta la cantidad de ancomicina liberada (g) desde la prótesis de acuerdo con el tramo de tiempo evaluado. En la figura 11, se presenta la gráfica representativa de esta cinética de liberación. De acuerdo con los resultados, en el tramo de la primera hora se observa la mayor liberación de vancomicina desde el implante. Desde la hora 2 en adelante, se observa que la liberación aumenta sostenida y paulatinamente con el avance del tiempo. Esta cinética de liberación permite en un inicio la exposición de una mayor cantidad del principio activo en la zona con el fin de alcanzar rápidamente concentraciones terapéuticas y luego la regulación de la salida del principio activo de forma paulatina en el tiempo. Si nos referimos a la tasa de liberación, en la primera hora se observa una alta tasa de liberación de vancomicina con un promedio de 218 g/mL, la que luego disminuye en el tramo de tiempo entre la primera y segunda hora, para luego aumentar y mantener la liberación sostenida. En la figura 12 se presenta la gráfica representativa de la tasa de liberación de vancomicina desde el implante en los tiempos de muestreo expuestos en la tabla 5. Tabla 5.- Cantidad de vancomicina liberada y tasa de liberación de ésta. A partir de los muestreos de los tiempos 2, 4, 8 y 12 horas se determinó la función o ecuación de la recta que permite predecir cuánto tiempo el implante seguirá liberando el principio activo: f (x) = 911, 6x+11.465, 1. Con esta información se extrapoló y determinó teóricamente la cantidad de vancomicina liberada en horas posteriores. En la tabla 6 se exponen las cantidades de vancomicina liberada acumulada y definida para los tiempos de 24, 36 y 43 horas. Esta simulación o considera que luego de las 12 horas, la velocidad podría disminuir, y por lo tanto, aumentar el tiempo de entrega. Tabla 6.- Cantidad de vancomicina liberada acumulada y determinada hasta las 43 horas. Ejemplo 7: Estabilidad principio activo en solución dentro del implante. Para este análisis se consideró nuevamente trabajar con el principio activo vancomicina, sin limitarse a otros principios activos. Con este ensayo se busca demostrar que la vancomicina es establece dentro del implante y no se degrada mientras es contenido, por lo tanto, su liberación es funcional. Para esto se analizó una muestra a tiempo cero (To) correspondiente a una muestra analizada inmediatamente luego de su preparación y una muestra que se dejó durante 24 horas en el implante. De esta forma, se compararon los resultados del tiempo 24 horas con el tiempo cero, como áreas de muestra y porcentaje de degradación a las 24 horas. La preparación de la solución muestra al tiempo cero (T0) consistió en tomar un frasco ampolla de vancomicina de 500 mg y adicionar 5 mL de suero fisiológico. Agitar la solución completa del liofilizado, tomar 0, 5mL de dicha ampolla, llevar a 5 mL de volumen final con suero fisiológico. Se toman estos 5mL y se inyectan en el depósito del implante. En el caso de la muestra a las 24 horas, se toma 0, 5 mL de una solución desde una ampolla de vancomicina de 500mg reconstituida en 5 mL de suero fisiológico. Los 0, 5 mL son llevados a 5mL de volumen final con buffer fosfato dibásico de potasio 0, 2 M pH 7, 4. Luego, con una jeringa se carga dicho volumen en el depósito o cápsula del implante, se sella el implante y se incorpora en un vaso precipitado vacío. La concentración nominal de la solución en la cámara es de 10 mg/mL. Luego de las 24 horas, se abrió la cápsula o depósito para extraer 0, 5 mL de la solución contenida en su interior. De los 0, 5 mL extraídos desde el depósito se toma una alícuota de 0, 1 mL que se llevan a un matraz de 100 mL para su análisis por HPLC con la misma curva de calibración y estándar descrito para el ejemplo anterior. En la tabla 7 se exponen los resultados de este ensayo. Se observa que a las 24 horas la degradación de la vancomicina no es significativa. Estos resultados demuestran que el principio activo incluido en el o los depósitos del implante de la presente invención se mantiene estable para su liberación. Tabla 7.- Resultados de ensayos de estabilidad de vancomicina en implante de la presente invención. Bibliografía (Kwarcinski, Jeremy & Boughton, Philip & Ruys, Andrew & Doolan, Alessandra & van Gelder, James. (2017) . Cranioplasty and Craniofacial Reconstruction: A Review of Implant Material, Manufacturing Method and Infection Risk. Applied Sciences. 7. 276. 10.3390/app7030276) . Chen Y, Zhang L, Qin T, Wang Z, Li Y, Gu B. (2019) . Evaluation of neurosurgical implant infection rates and associated pathogens: evidence from 1118 postoperative infections. Neurosurg Focus. 47 (2) :E6. doi: 10.3171/2019.5.FOCUS18582. Chengzhe Gao, Zongliang Wang, Zixue Jiao, Zhenxu Wu, Min Guo, Yu Wang, Jianguo Liu, Peibiao Zhang. (2021) . Enhancing antibacterial capability and osseointegration of olyetheretherketone (PEEK) implants by dual-functional surface modification, Materials & Design, Volume 205, 109733. https://doi.org/10.1016/j.matdes.2021.109733 Delaney, LJ, MacDonald, D., Leung, J., Fitzgerald, K., Sevit, AM, Eisenbrey, JR, Patel, N., Forsberg, F., Kepler, CK, Fang, T., Kurtz, SM, & Hickok, Nueva Jersey (2019) . Liberación de antibióticos activada por ultrasonido de los clips de PEEK para prevenir la infección por fusión espinal: evaluaciones iniciales. Acta biomaterialia, 93, 12-24. https://doi.org/10.1016/j.actbio.2019.02.041 Gu Xinming, Sun Xiaolin, Sun Yue, Wang Jia, Liu Yiping, Yu Kaixuan, Wang Yao, Zhou Yanmin. (2021) . Bioinspired Modifications of PEEK Implants for Bone Tissue Engineering. Frontiers in Bioengineering and Biotechnology, Vol. 8, doi:10.3389/fbioe.2020.631616. Hernandez Cantu, Enoc Isaí; Esparza Dávila, Sandra Paloma; Reyes Silva, Alan Karim Sayeg. (2020) . Eficacia de un modelo de prevención de infección de sitio quirúrgico en un hospital de segundo nivel de atención. Index Enferm, Granada, v. 29, n. 1-2, p. 9 12. Lazar, M. A., Vodnar, D., Prodan, D., Rotaru, H., Roman, C. R., Sorcoi, L. A., Baciut, G.; Campian, R. S. (2016) . Antibacterial coating on biocomposites for cranio-facial reconstruction.Clujul medical (1957) , 89 (3) , 430-434. https://doi.org/10.15386/cjmed-599 MINSAL. Ministerio de Salud de Chile. Departamento de Calidad y Seguridad en la atención, Programa de control de IAAS. (2017) . Informe de vigilancia de infecciones asociadas a la atención de salud. https://www.minsal.cl/wpcontent/uploads/2015/09/informe-vigilancia-2017.pdf Xue, Z., Wang, Z., Sun, A., Huang, J., Wu, W., Chen, M., et al. (2020) . Rapid construction of polyetheretherketone (PEEK) biological implants incorporated with brushite (CaHPO4.2H2O) and antibiotics for anti-infection and enhanced osseointegration. Mater. Sci. Eng. C Mater. Biol. Appl. 111:110782. doi: 10.1016/j.msec.2020.110782 Yan, J., Zhou, W., Jia, Z., Xiong, P., Li, Y., Wang, P., et al. (2018) . Endowing polyetheretherketone with synergistic bactericidal effects and improved osteogenic ability. Acta Biomater. 79, 216-229. doi: 10.1016/j.actbio.2018.08.037
Publicaciones:
ES2957984 (30/01/2024) - A1 Solicitud de patente con informe sobre el estado de la técnica
Eventos:
En fecha 20/01/2023 se realizó Registro Instancia de Solicitud
En fecha 20/01/2023 se realizó Admisión a Trámite
En fecha 20/01/2023 se realizó 1001P_Comunicación Admisión a Trámite
En fecha 23/01/2023 se realizó Superado examen de oficio
En fecha 21/11/2023 se realizó Realizado IET
En fecha 24/11/2023 se realizó 1109P_Comunicación Traslado del IET
En fecha 30/01/2024 se realizó Publicación Solicitud
En fecha 30/01/2024 se realizó Publicación Folleto Solicitud con IET (A1)
En fecha 18/04/2024 se realizó PETEX_Petición de examen sustantivo
En fecha 18/04/2024 se realizó PETEX_Petición de examen sustantivo
En fecha 18/04/2024 se realizó 5215P_Observaciones del solicitante al IET, Opinión Escrita y/o alegaciones a observaciones de terceros
Pagos:
20/01/2023 - Pago Tasas IET
+ ES-2957984_A11. Implante craneofacial de material PEEK para la liberación de sustancias o principios activos caracterizado porque comprende: Uno o más depósitos o recámaras interiores (1) ovaladas con paredes de 2, 5 a 4 mm de grosor, que almacenan un líquido o sustancia activa en formato líquido para ser liberado de forma gradual y prolongada al sitio del implante, tuberías (2) que interconectan los depósitos o recámaras interiores con los orificios de entrada y salida, definiéndose: a) una tubería de entrada que conecta el orifico de entrada (3) con el depósito (1) de tal forma que incluye un sistema de embudo que incluye una entrada o carga del líquido que tiene un diámetro de 1, 5-2, 0 mm y que luego se ensancha en una tubería de diámetro 2, 0 a 3, 0 mm para continuar con la tubería hasta el depósito. b) una tubería de salida que conecta el depósito o recámara (1) con el orificio de salida (4) que incluye un sistema de embudo de transición que presenta un diámetro de 07 a 2, 0 mm en la parte superior y un diámetro de 0, 7 a 1, 5 mm en su parte inferior, dependiendo del diámetro del orificio de salida (4) , Uno o más orificios de entrada (3) en la parte superior del implante dependiendo del número de depósitos, donde dicho orificio es la entrada o carga del líquido para llegar al o los depósitos (1) , donde el orificio de entrada tiene un diámetro de 1, 5-2, 0 mm y está bloqueado por un tapón de filamento PEEK o un tornillo de titanio, Uno o más orificios de salida (4) en la parte inferior del implante dependiendo del número de depósitos como orificio de salida gradual y prolongada del líquido o sustancia activa hacia el sitio del implante, donde el orificio de salida tiene un diámetro de 0, 7 a 1, 5mm y tiene un tapón o sistema que sella el implante para no permitir que el líquido se escape cuando no se ha dispuesto en el sitio del implante. 2. Implante craneofacial de material PEEK para la liberación de sustancias o principios activos de acuerdo con la reivindicación 1 caracterizado porque el orificio de entrada tiene un diámetro de 1, 5 mm. 3. Implante craneofacial de material PEEK para la liberación de sustancias o principios activos de acuerdo con la reivindicación 1 caracterizado porque el orificio de salida del líquido o sustancia activa tiene un diámetro de 0, 75mm. 4. Implante craneofacial de material PEEK para la liberación de sustancias o principios activos de acuerdo con la reivindicación 1 caracterizado porque el grosor de las paredes de los depósitos o recámaras es de 3, 5 mm. 5. Implante craneofacial de material PEEK para la liberación de sustancias o principios activos de acuerdo con la reivindicación 1 caracterizado porque las tuberías de todo el sistema tanto de entrada como de salida deben de ser de al menos 10 mm, con un máximo de 15 mm. 6. Uso del implante craneofacial de material PEEK para la liberación de sustancias o principios activos de acuerdo con las reivindicaciones 1 a 5 caracterizado porque sirve para la liberación prolongada y sostenida de sustancias y principios activos en formato líquido. 7. Uso del implante craneofacial de material PEEK para la liberación de sustancias o principios activos de acuerdo con la reivindicación 6 caracterizado porque sirve para la liberación prolongada y sostenida de al menos un principio activo soluble en un vehículo farmacéuticamente aceptable. 8. Uso del implante craneofacial de material PEEK para la liberación de sustancias o principios activos de acuerdo con la reivindicación 7 caracterizado porque sirve para la liberación prolongada y sostenida de principios activos analgésicos, antibióticos, antivirales, quimioterapéuticos, antiinflamatorios p cualquier principio activo soluble en un vehículo farmacéuticamente aceptable.
Los productos y servicios protegidos por este registro son:
A61F 2/28
Descripciones:
+ ES-2957984_A1 IMPLANTE CRANEOFACIAL DE POLIÉTER ÉTER CETONA (PEEK) CON DEPÓSITOS PARA ALMACENAR Y LIBERAR SUSTANCIAS ACTIVAS CAMPO DE LA INVENCIÓN La presente invención se relaciona con el área la biomedicina. Particularmente, se refiere a un implante craneofacial de material Poliéter éter cetona (PEEK) que dispone de depósitos para almacenar y liberar sustancias o principios activos en el sitio del implante. ANTECEDENTES DE LA INVENCIÓN En procedimientos quirúrgicos las infecciones son condiciones que con frecuencia son emergencias y que pueden determinar el fracaso o el éxito del procedimiento. En cirugías donde el procedimiento comprende la incorporación de prótesis, las complicaciones perioperatorias siguen siendo una preocupación importante. Una de las complicaciones más comunes y complejas son las infecciones bacterianas que pueden dar lugar a retrasos en la cicatrización, además de un aumento en los costos para el paciente e incluso la muerte (Chengzhe et al, 2021) . Este tipo de infecciones representan entre el 15 al 30% de las infecciones intrahospitalarias con una tasa de mortalidad entre 0, 6 y 1, 9% (Hernández et al., 2020) . La incidencia de infecciones posquirúrgicas depende del sitio y varía de <1 a 2% para reemplazos articulares y hasta 10% para cirugías de columna, además, estas tasas dependen de la salud del paciente y aumentan en pacientes de riesgo (ancianos, diabéticos e inmunodeprimidos) (Delaney et al., 2019) . En Chile, las infecciones de heridas operatorias se han mantenido como la tercera infección más frecuente, donde los indicadores de infección de herida operatoria son en pacientes con operaciones específicas, donde se incluyen las operaciones de colocación de prótesis (MINSAL, 2019) . En diversos estudios se ha revisado el fenómeno de infecciones post operatorias en el sitio del implante. En un estudio de revisión que incluye el metaanálisis de 227 estudios con 1118 pacientes que experimentaron infecciones, el ritmo de infección total observado fue de 4, 87%, lo que fue más severo en los procedimientos neuroquirúrgicos. Se identificó la presencia de bacterias grampositivos, como S. aureus, Staphylococcus coagulasa-negativo y P. acnes (Chen, Y., et al., 2019) . En el documento de revisión de Kwarcinski J. y colaboradores se destaca una amplia variación en el riesgo de infección informado para diferentes materiales en la reparación craneal. Se señala que los materiales seleccionados buscan imitar el hueso natural y ayudar a restaurar la función (estructural y estética) del cráneo humano pero que no están ausentes del riesgo de producir una infección. De acuerdo con los resultados de la revisión, 41 artículos que cumplían con los criterios de inclusión) , las tasas de infección promedio por material oscilaron entre 2, 04% y 10, 98%. Los resultados indican que existe variación entre los materiales con respecto al riesgo total de infección, sin embargo, dependiendo de los materiales comparados, este valor puede ser insignificante. Los factores de riesgo alternativos asociados con la infección, incluido el tiempo quirúrgico, las revisiones y la infección previa, tienen un mayor impacto en el potencial de infección que la variación material. La comparación de los métodos de fabricación destacó un efecto notable en la tasa de infección promedio. Se pueden observar tendencias que muestran que los materiales con mayores niveles de interacción superficial y soporte activo del crecimiento del tejido presentaron mayor resistencia a la infección. En el caso del material PEEK, las tasas de infección variaron entre 0, 00% y 14, 29%, con una tasa de infección promedio de 7, 89%, una desviación estándar de 5, 16% y un riesgo relativo de 0, 75 (Kwarcinski et a/., 2017) . Otra de las dificultades observada es la inflamación de la zona operatoria por la presencia del implante. Las prótesis se han ido actualizando de tal manera que se disminuyen los riesgos asociados a infecciones, por ejemplo, el cemento óseo se ha ido mejorando de manera tal que posea propiedades mecánicas y antibacterianas por lo que se disminuye el riesgo de infección. No así para las prótesis no cementadas, las cuales en la actualidad poseen características limitadas para prevenir o tratar infecciones bacterianas, donde básicamente la estrategia a realizar es cirugía de revisión (Chengzhe et a/, 2021) . El uso de materiales como PEEK son una alternativa como material para la fabricación de implantes en aplicaciones clínicas. Este material es biocompatible, químicamente estable radiotransparente y tiene un módulo elástico similar al del hueso natural (Gu et al., 2021) . Se han descrito estudios en donde se evalúa el uso de prótesis fabricadas con PEEK que pueden tener efectos antimicrobianos luego de ser colocados en el paciente para poder disminuir los riesgos de infección post operatorio. Por ejemplo, Delaney y colaboradores en 2019 describieron un depósito fabricado con PEEK que podía ser incorporado a un paciente como prótesis de columna. Esta prótesis podía liberar de forma prolongada antibióticos y/u otros compuestos, donde dicho depósito posee poros y al momento de aplicar ultrasonido los compuestos contenidos en el depósito son liberados de forma controlada y prolongada (Hernández et al., 2020) . Se han realizado estudios sobre prótesis generadas con PEEK con adiciones de nanopartículas o aleaciones con otros materiales que podrían tener actividad antimicrobiana. El documento de Yan y colaboradores en 2018 describe la elaboración de un recubrimiento de nanopartículas de plata y sulfato de gentamicina sobre una superficie porosa de PEEK (Yan et al., 2018) . El recubrimiento mejoró la capacidad bactericida del PEEK sobre bacterias Gram positivo y Gram negativo, además el número de bacterias adheridas a PEEK con el recubrimiento fue menor que en las condiciones de PEEK sin recubrimiento (Yan et al., 2018) . Xue y colaboradores en 2020 desarrollaron un método de deposición capa por capa (LBL) con ciclos controlados para construir rápidamente capas de brushita (CaHPO42H2O) (CaP) que contienen sulfato de gentamicina (GS) en PEEK para obtener PEEK modificado con CaP y GS (PEEK/CaP-GS) (Xue et al., 2020) . Los autores describen que realizaron ensayos antimicrobianos in vitro observando que las muestras de PEEK/CaP-GS tenían una propiedad antibacteriana excelente y sostenida, mientras que en ensayos de proliferación celular demostraron una compatibilidad aceptable y diferenciación osteogénica celular (Xue et al., 2020) . Por otro lado, el documento de Lazar y colaboradores en 2016, desarrolló un implante reforzado con fibra recubierto con gentamicina para reconstrucción craneofacial con propiedades antimicrobianas. Los resultados de este estudio indicaron que las bacterias fueron inactivadas al entrar en contacto directo con los recubrimientos de gentamicina (Lazar et al., 2016) . Chengzhe y colaboradores, en su documento publicado en 2021 presenta un estudio en el cual se desarrolló un implante PEEK que tiene por función combatir simultáneamente la contaminación bacteriana y promover la osteointegración mediante la liberación sostenida de clorhidrato de moxifloxacino (MOX) y péptido de crecimiento osteogénico (OGP) a partir de poli dopamina con superficie recubierta de PEEK sulfonado poroso (SPK) (Chengzhe et al, 2021) . Los resultados indican que la superficie SPK (SPD-MOX/OGP) modificada con MOX/OGP-PDA exhibió un efecto antibacteriano duradero y excelente contra Staphylococcus aureus planctónico/adherente y Escherichia coli in vitro. Además, se observó una mejora notable en la adhesión celular específica, la proliferación y la osteogenicidad en el sustrato SPD-MOX/OGP debido a la presencia de moléculas OGP y PDA en comparación con todos los demás grupos (Chengzhe et al, 2021) . En documentos de patente también se han descrito implantes para el control bacteriano en el sitio del implante. Por ejemplo, el documento CN101432030B describe un cuerpo o depósito tridimensional que incluye una o más sustancias implantables. Dicho deposito puede recibir cantidades de un líquido. El depósito más el líquido pueden formar una masilla que puede ser útil como material implantable. Otro documento como WO2007001624A2 divulga un dispositivo médico implantable para el tratamiento de la osteonecrosis. Dicho dispositivo de implante adaptado para su inserción en uno o más canales o huecos en el tejido óseo; una pluralidad de depósitos discretos ubicados en la superficie del al menos un cuerpo del dispositivo de implante; y al menos un sistema de liberación dispuesto en uno o más de la pluralidad de depósitos, en el que el sistema de liberación incluye al menos un fármaco seleccionado del grupo que consiste en promotores del crecimiento óseo, promotores de la angiogénesis, analgésicos, anestésicos, antibióticos y combinaciones de los mismos. En el documento US20170056565A1 se divulga un clip biocompatible fabricado con PEEK para el tratamiento de infecciones sitio quirúrgicos, el cual va adherido a un implante. Este clip comprende un depósito y una abertura del depósito, donde esta última está sellada con un material sensible a la presión que puede romperse al aplicar una fuerza a dicho material y liberar de forma prolongada compuestos. El depósito puede ser llenado con un agente terapéutico seleccionado del grupo que consiste en: un antibiótico, antivírico, analgésico, factores de crecimiento, antifúngico, antimicobacterias, quimioterapéutico, osteogénico, como ascorbato para aumentar la ormación de hueso, hormona paratiroidea, péptidos, peptoides, AINE, analgésicos o combinaciones de los mismos. El documento US8821912B2 describe métodos para fabricar dispositivos médicos implantables, que pueden ser fabricados con PEEK y poseen propiedades antimicrobianas. El efecto antimicrobiano de estos dispositivos se produce por la incorporación de partículas cerámicas con cationes metálicos antimicrobianos en resina PEEK fundida, que posteriormente se deja enfriar y fraguar en su forma final lograda mediante moldeo por inyección, corte y mecanizado u otras técnicas. Si bien, en el estado del arte se han descrito y realizado avances en la fabricación de prótesis con propiedades antimicrobianas, aun se requieren alternativas mejores para evitar la generación de infecciones en el sitio del implante de forma prolongada y controlada. Es reconocido en la práctica clínica que las primeras 48 a 72 horas de la cirugía de incorporación del implante son vitales por lo que se requieren alternativas de implante que sean capaces de mantener un ambiente antimicrobiano prolongado durante este tiempo para evitar la generación de infecciones. Las propuestas existentes hasta ahora corresponden a implantes con acción antimicrobiana de contacto y momentánea, o que requieren la funcionalización con nanopartículas, lo que complica técnicamente y encarece el proceso de producción del implante. En otros casos, se acoplan clips o dispositivos externos en la superficie del implante, esto conlleva la complicación del proceso de producción y el ajuste espacial del implante complejo en el cráneo del paciente. Por lo tanto, existe la necesidad de un implante craneofacial que pueda liberar de forma constante y prolongada sustancias activas de interés e importancia clínica en la zona del implante. Esto permitiría en ciertos casos, ejercer o promover un ambiente antimicrobiano prolongado y controlado durante las primeras horas de fijación del implante para evitar la generación de infecciones, y que, a su vez, sea un implante simple y de menor costo de producir y de trabajar. En la presente solicitud se describe un implante, particularmente un implante craneofacial fabricado en PEEK, el cual comprende o incluye uno o más depósitos o ecámaras de almacenamiento que en su interior pueden almacenar principios activos o sustancias en estado líquido o formato líquido que son liberados de forma controlada y gradual por gravedad en el sector del reemplazo óseo. DESCRIPCIÓN DE LA INVENCIÓN La invención corresponde a un implante craneofacial de material Poliéter éter cetona (PEEK) para la liberación de principios o sustancias activas, porque dicho implante comprende: Uno o más depósitos o recámaras o cápsula interiores (1) ovaladas con paredes de 2, 5 a 4 mm de grosor, que almacenan un líquido o sustancia activa en formato líquido para ser liberado de forma gradual y prolongada al sitio del implante, tuberías (2) que interconectan los depósitos o recámaras interiores con los orificios de entrada y salida, definiéndose: a) una tubería de entrada que conecta el orifico de entrada (3) con el depósito (1) de tal forma que incluye un sistema de embudo que incluye una entrada o carga del líquido que tiene un diámetro de 1, 5 a 2, 0 mm y que luego se ensancha en una tubería de diámetro 2, 0 a 3, 0 mm para continuar con la tubería hasta el depósito. b) una tubería de salida que conecta el depósito o recámara (1) con el orificio de salida (4) que incluye un sistema de embudo de transición que presenta un diámetro de 0, 7 a 2, 0 mm en la parte superior y un diámetro de 0, 7 a 1, 5 mm en su parte inferior, dependiendo del diámetro del orificio de salida (4) , c) Uno o más orificios de entrada (3) en la parte superior del implante dependiendo del número de depósitos, donde dicho orificio es la entrada o carga del líquido para llegar a los depósitos (1) , donde el orificio de entrada tiene un diámetro de 1, 5 a 2, 0 mm y está bloqueado por un tapón de filamento PEEK o un tornillo de titanio, Uno o más orificios de salida (4) en la parte inferior del implante dependiendo del número de depósitos como orificio de salida gradual y prolongada del líquido o sustancia activa hacia el sitio del implante, donde el orificio de salida tiene un diámetro de 0, 7 a 1, 5mm y tiene un tapón o sistema que sella el implante para no permitir que el líquido se escape cuando no se ha dispuesto en el sitio del implante. El implante craneofacial de la presente invención evita las infecciones post-implante, particularmente, las infecciones bacterianas. Este efecto lo logra gracias a la liberación sostenida, controlada y prolongada de un líquido, sustancia o principio activos en formato líquido. Los inventores han definido específicamente el diseño y ubicación espacial de cada componente dentro del implante de tal manera de disponer de depósitos o recámaras lo suficientemente grandes para acumular una cantidad considerable de un líquido o principio activo o sustancia activa en esta forma, pero al mismo tiempo, que el implante no pierda sus características y propiedades estructurales. Los inventores llevaron a cabo una serie de iteraciones experimentales para definir las mejores condiciones estructurales de tamaño, longitud y diámetro de cada parte o componente del implante craneofacial de la presente invención. Fue un desafío técnico y de diseño el generar un implante con uno o más depósitos o sistemas de recámaras, más bien multicámaras de almacenaje interno para líquido con una capacidad apropiada. Más complejo e importante, cómo se añade y libera el líquido de interés desde el implante al paciente. Así, en las iteraciones se definió empíricamente que el depósito puede contener uno o más depósitos o sistemas de recámaras de almacenamiento dependiendo de la necesidad de vaciado del líquido a almacenar. Particularmente, se definieron implantes con un depósito de capacidad de 5, 0 mL de llenado e implantes con 2 o más depósitos para incluir al menos 2, 5 mL del principio activo o sustancia en formato o estado líquido. Aun cuando inicialmente se observó que era difícil manejar el flujo del líquido y las proporciones de los orificios de entrada y salida para lograr la salida controlada y prolongada en los implantes con múltiples o más cámaras, los inventores incluyeron en sus iteraciones experimentales la definición de cada diámetro de entrada y salida, en onjunto con las longitudes y disposición física de los componentes de cierre (tapón) que impiden la salida del líquido cuando aun no se ha utilizado el implante y permiten su correcto funcionamiento. Cuando los inventores evaluaron los diámetros de los orificios de entrada y salida, también se encontraron con diferentes problemáticas para lograr la liberación correcta y prolongada del líquido o principio activo depositado en las recámaras. En la presente invención las condiciones de los tamaños de depósito o cápsula y de orificios de entrada y salida guardan relación con la regulación de la salida o liberación del líquido o sustancia activa en formato líquido desde los depósitos o cápsulas al medio. Como parte de las iteraciones experimentales, los inventores definieron que los orificios de entrada en la parte superior del implante para la entrada o carga del líquido o sustancia activa, deben tener un diámetro apropiado para que se pueda acoplar una jeringa que introduzca dicho principio activo o líquido al implante. A su vez, el orificio no debe colapsar ni permitir la salida o sobrepaso del líquido. En ese sentido, los inventores han definido que la tubería de entrada que conecta el orifico de entrada (3) con el depósito (1) incluye un sistema de embudo que incluye una entrada o carga del líquido que tiene un diámetro de 1, 5-2, 0 mm y que luego se ensancha en una tubería de diámetro 2, 0 a 3, 0 mm para continuar con la tubería hasta el depósito, donde el orificio de entrada tiene un diámetro de 1, 5-2, 0 mm. Como parte de las mejoras se adicionó también una sección adicional en el orificio de entrada del implante para insertar un trozo de filamento y que funcione como tapón. Este tapón puede ser de material PEEK o puede corresponder a un tornillo de titanio. En el caso de los orificios de salida en la parte inferior del implante como orificio de salida gradual y prolongada del líquido o sustancia activa hacia el sitio del implante, los inventores observaron en las pruebas expuestas en los ejemplos de aplicación la necesidad de regular la salida del líquido desde los depósitos o cápsulas de tal forma de aumentar el tiempo efectivo de liberación y exposición el líquido en el ambiente o sector del implante. Es importante destacar que los inventores han definido todas las condiciones de sistema tipo embudo, diámetros de entrada y salida y las distancias y grosores mínimos de cada estructura y disposición de éstas en el diseño del implante, de tal forma de permitir la liberación prolongada de la sustancia activa en formato líquido. Así, los inventores definieron que el implante debe disponer de uno o más orificios de salida (4) en la parte inferior del implante dependiendo del número de depósitos como orificio de salida gradual y prolongada del líquido o sustancia activa en formato líquido hacia el sitio del implante, donde el orificio de salida tiene un diámetro de 0, 7 a 1, 5mm y tiene un tapón o sistema que sella el implante para no permitir que el líquido se escape cuando no se ha dispuesto en el sitio del implante. La regulación del tamaño del diámetro del orificio de salida y los tamaños definidos por los inventores para cada estructura del implante permiten la liberación gradual del líquido o sustancia activa den formato líquido. En el caso de las tuberías que se interconectan a los depósitos o recámaras, se disponen de tuberías en la parte superior de los depósitos o recamaras que originan los orificios de entrada para la incorporación del líquido a dichos depósitos y tuberías que van desde los depósitos a la zona inferior o parte de abajo del implante y que dan origen a los orificios de salida con diámetros controlados y definidos según lo ya descrito. Las tuberías de todo el sistema tanto de entrada como de salida deben de ser de al menos 10 mm, con un máximo de 15 mm, este mínimo se debe a las pruebas desarrolladas para que el líquido pueda entrar de manera adecuada a la recámara o depósito. En una de las formas de la invención, la liberación del líquido o sustancia activa en formato líquido se relaciona directamente con la entrega de sustancias activas en el sitio del implante, luego de su inclusión en el cuerpo del paciente. En ese sentido, la liberación de sustancias activas en formato líquido desde el implante al sitio de implante permite generar un efecto específico y prolongado en el lugar del implante luego de la cirugía. En una de las formas de la invención, la liberación prolongada y sostenida durante los primeros días luego del implante permite evitar y disminuir infecciones del tipo postoperatorias. La liberación prolongada de sustancias activas las primeras horas postoperación, como sustancias antibióticas y antiinflamatorias, evita la colonización de bacterias que pueden causar alguna infección en el sitio del implante. Tal como se demuestra en los ejemplos de aplicación, en particular en la figura 1 parte del ejemplo de aplicación 1, el implante libera por goteo. Los inventores definieron los tamaños específicos para la liberación por goteo como un mecanismo de liberación prolongada en la zona del implante. En una de las formas de la invención, la liberación de sustancias o principios activos en formato líquido desde el implante permite un efecto clínico local dependiendo del tipo de principio activo, sin limitarse, a antibióticos, antiinflamatorios, analgésicos u otro tipo de principio activo. Se incluyen también como principio activo agentes quimioterapéuticos o cualquier principio activo soluble que pueda conformar una solución. Como parte del alcance de la invención, se puede incluir al menos un principio activo, siendo posible poder incluir más de uno, donde estos se pueden incluir en el mismo depósito o en depósitos separados. Los inventores han demostrado en los ejemplos de aplicación 2 y 3 que los implantes parte del alcance de la invención pueden liberar de forma prolongada un líquido o sustancia activa en formato líquido desde uno o más depósitos o recámaras o cápsulas incluidas en la estructura interna del implante. La disposición física, espacial y los tamaños de cada componente del implante permiten la liberación de la sustancia líquida desde las 24h, hasta incluso 11 días luego de incluir en él algún tipo de sustancia en este formato. Incluso, en condiciones simuladas de movimiento (simulación del movimiento de la cabeza del paciente) se apreció la liberación sostenida y prolongada de la sustancia líquida desde el implante al medio, por varios días. Estos resultados demuestran que el implante parte de la invención es funcional y libera de forma sostenida y prolongada el líquido o sustancia en formato líquido incluida en él o los depósitos del implante. Es importante destacar que los resultados expuestos en los ejemplos de aplicación 1 a 6, en particular para los ejemplos 5 y 6, si bien fueron evaluados para vancomicina como principio activo, la prueba y resultados es extrapolable y válida para cualquier otro principio activo que se pueda solubilizar en algún solvente apropiado, en este caso solventes o vehículos farmacéuticamente aceptables. Un ejemplo de solvente puede ser agua, suero fisiológico, buffer fosfato dibásico de potasio, buffer fosfato salino o cualquier otro buffer o vehículo aceptable para su administración a un ser vivo, en particular a un ser humano. En ese sentido, es parte del alcance de la invención la posibilidad de incluir cualquier tipo de principio activo que sea soluble en un vehículo farmacéuticamente aceptable según lo ya señalado. En el ejemplo 5 se expone el análisis de la concentración y tasa de liberación. La cantidad de vancomicina liberada en la primera hora fue de 13.087, 5 g. Luego en la hora 2 se observa una diminución de la cantidad liberada con 548, 5 g pero que luego se retoma o aumenta en la hora 4 con 1.232, 5 g del principio activo. Para las horas 8 y 12 del ensayo se observó también un aumento con 3.505, 5 g y 4.309, 4 g, respectivamente. Respecto de la tasa de liberación, en la primera hora se observa una alta tasa de liberación de vancomicina con un promedio de 218 g/mL, la que luego disminuye en el tramo de tiempo entre la primera y segunda hora, para luego aumentar y mantener la liberación sostenida. Esta cinética de liberación permite indicar que primero hay una liberación rápida o mayor del principio activo para luego mantener una liberación paulatina y constante. En el ejemplo de aplicación 6 se demuestra que el principio activo cargado en el o los depósitos del implante se mantiene estable y no pierde su estructura ni se degrada, por lo que es funcional. En el caso particular, se evaluó la estabilidad de una solución de vancomicina en el implante por 24 horas, observándose que no existe degradación significativa del principio activo por lo que el depósito no afecta a la estabilidad del principio activo a liberar. Esta información permite señalar que el principio activo se libera de forma correcta y con una cinética de liberación apropiada para la liberación de principios activos o fármacos en la zona del implante craneofacial, donde se observa primero la salida rápida para lograr el efecto terapéutico, una disminución y luego la mantención de la liberación por al menos las primeras 43-48 horas. Además, la puesta del principio activo o solución de éste en el depósito del implante no afecta su estabilidad. El implante descrito en la presente invención corresponde a un implante craneofacial fabricado mediante impresión 3D en material PEEK customizado. Dicho implante comprende dos orificios o aberturas, una superior y una inferior que permiten el llenado y salida de las sustancias a liberar, respectivamente, tal como se ha descrito para la presente invención. En las realizaciones de la invención, los receptáculos o depósitos o cámaras o reservorios descritos en el implante, pueden incorporar sustancias o medicamentos o principios activos en formato o estado líquido, los cuales pueden ser liberados en el sector donde se realizó el reemplazo óseo. En realizaciones de la invención, los implantes con depósitos o recámaras o reservorios incorporan particularmente medicamentos o principios como antibióticos y/o antiinflamatorios y/o analgésico con el fin de llevar al éxito la incorporación de dicho implante. Es también parte del alcance de la invención que el medicamento o principio activo corresponda a un agente quimioterapéutico o cualquier otra sustancia que se pueda disolver y obtener como una solución líquida. Es importante también señalar que debido a que el implante puede tener más de un depósito, también es parte del alance de la invención La liberación de sustancias a través del receptáculo, depósito cámara o reservorio del implante customizado con PEEK, descrito en la presente invención se debe a la acción de la gravedad, la cual permite la liberación gradual de dichos compuestos. La liberación depende del diámetro del orificio de salida y demás tamaños de las estructuras del implante. Dentro de las realizaciones de la invención, el desarrollo del implante customizado incluye depósitos o recamaras interiores y con un sistema interconectado de tuberías, donde dichas recamaras pueden almacenar sustancias liquidas las cuales pueden ir siendo liberadas por la fuerza de gravedad. Particularmente, se presenta un implante con depósitos o recamaras superiores e inferiores que permiten el paso por gravedad de las sustancias, donde dichas sustancias pueden ser antibióticos y/o antiinflamatorios y/o analgésicos. En realizaciones de la invención, se describe que las sustancias liquidas que pueden ser incorporadas en las recamaras internas del implante mediante el uso de una jeringa quirúrgica. Definiciones El presente documento describe un implante craneofacial, donde este último se refiere a cualquier implante que puede ser colocado en un paciente en las zonas del cráneo y/o cara. El implante craneofacial de la presente invención permite la liberación prolongada y sostenida de principios activos o sustancias activas, particularmente en forma líquido, para generar un efecto específico terapéutico en la zona del implante. Se incluyen diferentes tipos de sustancias o principios activos en busca de efectos de analgesia, antinflamatorio u antibiótico, agentes quimioterapéuticos o cualquier principio activo capaz de incluirse en una solución. En una de las formas de la invención, permite evitar las infecciones post-implante, particularmente, las infecciones bacterianas. Una medida de esto es la diminución del recuento bacteriano. En ese sentido, cuando se ha referencia a la disminución del recuento bacteriano, se hace referencia a la disminución en el recuento de UFC o UFC/mL de las bacterias. Cuando en el documento se hace referencia a PEEK customizado, se indica que el implante descrito en la presente invención es fabricado en PEEK y es personalizado según las características del paciente que recibirá el implante. Cuando en el documento se indica el término "receptáculo", "cámara", "deposito", "reservorio" y/o "recamara interior" y/o "cápsula" se indica una cavidad en que se contiene o puede contener una sustancia en su interior. El término "postoperatorio" se refiere al periodo que sigue luego de una intervención quirúrgica y que finaliza con la rehabilitación del paciente. El término "perioperatorio" corresponde al periodo de tiempo de un procedimiento quirúrgico, el cual comprende desde la preparación del paciente hasta su recuperación. Cuando en el documento se indica "liquido activo", "principio activo" o "sustancia activa" se hace referencia a toda sustancia o mezcla de estas que poseen una función y/o actividad particular en la composición de un medicamento. En el documento se indica el término "iteraciones", este término hace referencia a una repetición de pasos para un determinado fin. En el caso de la presente invención, las iteraciones corresponden a la repetición de la fabricación del implante y las realizaciones e pruebas sobre este para llegar a obtener el implante con la funcionalidad que se requiere. Descripción Figuras Figura 1.- Imagen prototipo iteración 1 para desarrollo de implante. Se presenta el prototipo 1 compuesto de múltiples cámaras de depósito. Fallo de goteo y salida del líquido desde el reservorio. A) presenta la estructura del prototipo de implante; b) se describen las partes del prototipo, donde (1) recamaras de contención del líquido o principio activo con capacidad total de 3 mL, (3) entrada del líquido o principio activo mediante jeringa con diámetros de 0, 8, 1, 25 y 1, 25 mm; y (4) orificios de salidas con diámetro de 0, 5, 0, 7 y 1 mm para el líquido o principio activo. Figura 2.- Imagen prototipo iteración 2 para desarrollo de implante. En A) Se presenta el Prototipo 2 de implante con una sola recámara o depósito (1) . B) de describe la entrada del líquido o principio activo por una tubería de 26 mm de largo (2) y el espesor del prototipo de 4, 7 mm se indica con una flecha negra gruesa. Prototipo 2 falló pues el implante pierde capacidades mecánicas, al debilitarse la estructura interna del implante. Figura 3.- Imagen prototipo de iteración 3 para desarrollo de implante. Implante prototipo 3 con 2 recámaras independientes para el depósito de líquido o principio activo. Prototipo fallido por problemas en el diámetro de salida de la tubería de uno de los depósitos. En (1) se indican los depósitos de líquido o principio activo; (3) orificios de entrada del líquido o principio activo por medio de una jeringa; (4) orificios de salida del líquido o principio activo. Figura 4.- Imagen prototipo de iteración 4 para desarrollo del implante. Se agrega material en la parte interna para tener al menos 2, 5 mm de grosor en cada lado de la recámara o depósito (1) . En (2) diámetro de tubería de 1, 4 mm, (3) el orificio de entrada para un diámetro de jeringa de 1, 5 mm, (4) orificio de salida de diámetro de salida de 0, 75 mm, o 0, 5 mm. Figura 5.- Imagen de iteración 5 para desarrollo del implante. Se provee un implante con 2 cámaras o depósitos internos con pares de separación de 3, 5 mm de grosor y con os orificios de entrada y salida de líquido definidas. A) esquema digital del implante, b) molde del implante. Figura 6.- Imagen de iteración 6 de mejora del implante: tapón en orificio tubería de ingreso del líquido al implante y sistema tipo embudo. En A) se presenta una imagen de la parte superior del implante donde se aprecia la tubería de entrada y el orificio de entrada donde se dispone un trozo o pieza de PEEK para funcionar como un tapón que impide que el líquido contenido en el interior del implante se escape o devuelva por el orificio de entrada. En B) se presenta un implante que incorpora mejoras como un sistema tipo embudo en el ingreso del líquido, este sistema va variando los diámetros de cada cañería según lo que se muestra en la imagen con 1, 5 mm para la entrada de la aguja de la jeringa que incorpora el líquido al implante y luego un ensanche en una tubería de 3, 0 mm en la tubería hacia el depósito o recámara. En dicho implante, el orificio de entrada expone una tubería de salida con un sistema embudo que tiene dimensiones de 2, 0 mm para luego disminuir su diámetro a 1, 3 mm en el orificio de salida. El implante de la imagen tiene un solo depósito de 5 mL de capacidad con paredes de un grosor de 2, 5 mm de espesor. En 6C) se expone el tapón o sistema que sella el implante en el orificio de salida para no permitir que el líquido se escape cuando no está en uso (ver figura 6C) . Figura 7.- Imagen de iteración 6 de mejora del implante: tapón en orificio tubería de ingreso del líquido al implante y sistema tipo embudo en salida. En A) Se expone el prototipo de implante con un depósito de medidas 52, 2 mm de ancho y 52, 86 mm de alto para el depósito. En B) se expone un prototipo de implante de dos depósitos o recámaras que presenta una recámara izquierda de 24, 43 mm de ancho por 46, 7 mm de alto y un recámara o depósito izquierdo con un ancho de 24, 47 mm y 46, 84 mm de alto. Figura 8.- Fotografía Ensayo de liberación de líquido desde un implante de la presente invención (ejemplo de aplicación 2) . Se observan las tonalidades diferentes de azul de acuerdo con la liberación del azul de metileno diluido en agua cuando el implante se dispone en un recipiente. Se valuaron de izquierda a derecha los tiempos de liberación de 1, 2, 3, 4 y6 horas. Figura 9.- Fotografía Prueba de verificación del tiempo de vaciado en implante de un depósito (ejemplo de aplicación 3) . En A) se expone una fotografía de los resultados de la observación de las muestras obtenidas en los puntos de tiempo de 24, 72, 96, 120 y 144 horas. De izquierda a derecha, en el tiempo de 24 horas se observó muy poca coloración del medio o salida del líquido desde el implante. En horas intermedias de 72, 96 y 120 horas se observa mayor liberación del líquido con una tonalidad celeste suave observada en el ambiente (recipiente) para que a las 144 horas la tonalidad cambie a una tonalidad celeste fuerte. Cuando se evaluó el tiempo de 168 horas se observa una mucho mayor liberación del contenido desde el reservorio o cápsula dentro del implante al medio (recipiente) . En B) se expone una fotografía del orificio de salida o liberación del líquido contenido en el implante, en este caso azul de metileno diluido en agua. Se observa la salida del líquido desde el orifico de salida del implante con una coloración azul más fuerte. Figura 10.- Imagen implante con depósitos y tuberías para la liberación gradual de un líquido o principio activo al cerebro. En A y en B se presenta la imagen del implante integrado en el cráneo del paciente. En A, se observa el detalle del interior del implante. En B, el implante es visto desde la parte interna del cráneo, en donde las salidas de las recamaras liberan hacia el interior del cerebro el líquido. Figura 11.- Gráfica vancomicina liberada (g) desde la prótesis de acuerdo con el tramo de tiempo evaluado. Se presenta la gráfica representativa de esta cinética de liberación considerando los g liberados para los puntos de muestreo de 0, 2, 4, 8 y 12 horas. Figura 12.- Tasa de liberación de vancomicina desde el implante por tiempo de muestreo. Se expone la gráfica de la liberación de g/min de acuerdo con el tiempo de muestreo evaluados de 0, 2, 4, 8 y 12 horas. Ejemplos de aplicación Ejemplo 1: Desarrollo de prototipos de implantes con depósitos y tuberías para liberación controlada de líquidos o sustancias activas Es parte del presente ejemplo de aplicación, el desarrollo del prototipo de implante considerando las iteraciones y análisis experimentales correspondientes para obtener n implante que conste de depósitos o recamaras interiores interconectados con tuberías de tal forma de poder incluirse en él, un líquido o sustancia activa, pero también que se libere de forma prolongada durante las primeras 24-48 horas luego de incorporar el implante al paciente. Para las pruebas se utilizó un implante 3D previamente diseñado, al cual se le adicionó uno o más depósitos o recámaras de almacenaje interno de líquido. - Primera iteración de prueba: Implante con más de un depósito o sistema de recámaras El primer prototipo de implante que se desarrolló contaba con varios depósitos o recamaras interiores y con un sistema interconectado de tuberías, con el fin que cada recamara almacenara líquido (prueba con antibiótico) y por gravedad se fuera trasladando este líquido desde las recamaras con mayor cantidad a las de menor cantidad. Este prototipo contaba con tres entradas de líquido a través de una cañería recta, con un diámetro de 0, 8 mm; 1, 25 mm y de 1, 5 mm. En ese sentido, los inventores observaron que el diámetro de entrada es de suma importancia pues debe ser lo suficientemente grande para permitir cargar correctamente el líquido al interior con una jeringa, pero al mismo tiempo, no ser tan grande para que en el proceso de carga el líquido no rebote y se salga del implante. De acuerdo con las pruebas de este prototipo, el mejor diámetro fue de 1, 5 mm. Este primer prototipo funcionó respecto de la salida o liberación de líquido pues se observaron varias salidas de líquido por goteo, pero al ser un sistema de cañerías tan extenso y con diámetros delgados ocurrió que algunas de estas salidas no funcionaron. Las salidas evaluadas fueron de diámetros 0, 5 mm; 0, 7 mm y 1 mm. En este caso aun cuando el implante no funcionó por completo de forma correcta, se observó que la salida de 0, 7 mm permite la salida controlada de goteo. La salida de 0, 5 mm se obstruía y la salida de 1 mm presentaba un flujo muy alto de salida (figura 1A y 1B) . En la tabla 1 se resumen los resultados de la definición de los diámetros de los orificios de entrada y salida del líquido a incluir en el implante para ser liberado al sector de inserción del implante. Tabla 1. Resultados determinación orificios de entrada y salida del implante. - Segunda iteración de prueba: implante con un depósito o recámara Para las pruebas se utilizó un implante 3D previamente diseñado, al cual se le debió adicionar un depósito o sistema de almacenaje interno de líquido. En primer lugar, se diseñó el sistema de cañerías y la forma de unión o interacción con el depósito o recámara. Se proveyó una entrada de líquido que se dirige hacia la recámara y con una salida que se encuentra sobre él, esta salida posee una forma piramidal para poder controlar la velocidad y cantidad de líquido que se deposita. Luego, otro de los desafíos de diseño fue definir la capacidad interna del depósito o recámara. La primera prueba se realizó con un prototipo de implante que incluyó un depósito de capacidad interna de 2 mL. Se produjo un implante de un tamaño de 85, 62 mm x 99, 7 mm con un espesor de 4, 7 mm, compuesto de una sola recámara o depósito con una tubería de 26 mm de largo conectada a él (ver figura 2A y 2B) . Para evaluar el funcionamiento del implante, en particular en la liberación de un líquido o principio activo, se realizó una prueba adicionando agua líquida al implante por medio de una jeringa. Se observó que el depósito único de 2 mL es muy grande pues el implante pierde capacidades mecánicas, al debilitar la estructura interna del implante. Para solucionar esta problemática y que el implante presente propiedades mecánicas adecuadas, se estableció en el diseño que las paredes de cada depósito deben tener al menos 2, 5 mm de grosor. Cuando se probaron implantes con estas consideraciones, los implantes mantenían sus propiedades mecánicas. - Tercera iteración de prueba: Mejora implante con más de un depósito o recámaras Se realizó un segundo prototipo o mejora de los implantes con dos depósitos o recamaras independientes, cada una de estos dispone de una entrada de líquido y una salida. Cada recámara tiene una capacidad de almacenamiento de al menos 2 mL, por lo que la suma de las capacidades de las recamaras puede llegar a hacer de alrededor de 5 mL. El implante utilizado es alrededor de 20% más grande que el anterior, pues se requiere una mayor superficie para tener mayor capacidad de almacenamiento de líquido y no disminuir las propiedades mecánicas del implante. El implante comprende una tubería conectada a cada depósito, donde dicha tubería tiene una entrada del diámetro de una jeringa de 1, 2 mm. El implante tiene además unas tuberías de salida de un diámetro de 0, 5 mm para un depósito y de 0, 35 mm para el otro depósito (figura 3) . La prueba en este caso fue parcialmente exitosa ya que solo una de las dos recamaras cumple el objetivo de dejar fluir el líquido en forma de goteo, la otra entrada al ser de un tamaño tan pequeño por el método de fabricación se encuentra totalmente tapada. - Cuarta iteración de prueba La tercera iteración se basa en el mismo prototipo de implante anterior, pero con una gran diferencia, se agrega material en la parte interna para tener al menos 2, 5 mm de grosor en cada lado de la recámara o depósito. Esto de acuerdo con lo observado en la egunda iteración de prueba, mejora las condiciones y propiedades mecánicas del implante. Como parte de este prototipo, se aumentó el diámetro de la tubería de salida del líquido, pero para poder lograr el efecto y que sea una liberación controlada, en la parte superior se incluyó un sistema de embudo (figura 4) . Este implante tiene dos diámetros de salida diferente, de 0, 75 mm y 0, 5 mm. Se observó que el mejor diámetro de salida es de 0, 75 mm. En el caso de los 0, 5 mm el líquido no fluye de forma simple por lo que es difícil controlar el flujo del líquido. Al realizar una incorporación de material en las zonas de las recamaras se logró que el implante no disminuya sus capacidades mecánicas, logrando obtener una excelente capacidad de almacenaje en ambas recámaras sin afectar la calidad e integridad del implante. - Quinta iteración de prueba En una iteración posterior del prototipo previo, se procedió a disponer las recamaras de almacenamiento en la parte interior del implante conservando al menos 3, 5 mm de grosor entre la recamara y la parte exterior del implante. Para obtener el mismo grosor en la parte interior del implante es que se realizó un proceso de engrosamiento de la recamara, así obtener al menos 3, 5 mm de grosor, esta prueba resulto un completo éxito (figura 5) . Por lo tanto, el implante puede comprender solo un depósito de almacenamiento el que comprende paredes de un grosor de 2, 5-4 mm., preferentemente de 3, 5 mm. - Sexta iteración de prueba: Definición final sistemas de entrada y salida del líquido Considerando todas las problemáticas observadas en las iteraciones previas respecto al control de entrada y salida del líquido a liberar en el lugar el implante, se realizaron diversas mejoras al implante prototipo. Tapón orificio en tubería de entrada del líquido a almacenar En la sección inicial de la tubería por donde entra el líquido se adicionó al implante un trozo o pieza de la misma materia prima con la que se desarrolla el implante (PEEK) para funcionar como un tapón que impide que el líquido contenido en el interior del implante se escape o devuelva por la entrada. El filamento utilizado tiene un diámetro de 1, 75 mm, pero los termoplásticos tienen una dilatación por ello la entrada es 0, 25 mm mayor que la del filamento. Por lo tanto, el diámetro adecuado es de 2, 0 mm para bloquear la salida con el filamento (ver figura 6A) . Cuando se instala el implante, este trozo o filamento puede ser eliminado fácilmente (remover, quitar, traccionar) y una vez dispuesto el implante en su sitio, esta apertura queda cubierta por el hueso de la zona del implante. En este caso también es posible técnicamente incluir un tornillo de titanio de 1.5 mm que se enrosca en la salida para ser también un tapón o sello. Sistema embudo en la tubería de ingreso del líquido Se incorporó un sistema tipo embudo en el ingreso del líquido, este sistema va variando los diámetros de cada cañería, tiene como finalidad lograr que el líquido en el interior no se devuelva. Es un segundo método para impedir que el líquido se escapa del sistema de tuberías por el orificio de entrada. El sistema embudo conlleva que la tubería tenga un diámetro de 1, 5 mm para la entrada de la aguja de la jeringa que incorpora el líquido al implante y luego se ensancha en una tubería de 3, 0 mm. Es decir, existe un punto de embudo o transición para que el líquido no se devuelva (ver figura 6B) . Sistema de salida en la tubería de salida del líquido hacia el sitio del implante Se mejoró el sistema de salida, definiéndose que el diámetro del orificio de salida del líquido puede ir desde los 0, 7 mm hasta 1, 5 mm dependiendo de la geometría del implante. En este nuevo prototipo la tubería final de salida del líquido incorpora un sistema tipo embudo que busca favorecer que el vaciado de la recamara sea aún más controlada y prolongada (ver figura 6B) . Además, en esta prueba se adicionó un tapón o sistema que sella el implante para no permitir que el líquido se escape (ver figura 6C) , una vez que el implante se encuentra en pabellón, el equipo quirúrgico debe cortar una parte del cilindro donde quedará expuesto el orificio de salida para que el líquido pueda fluir desde el implante al interior del cuerpo. Medidas generales implantes y depósitos En cuanto a los tamaños, el implante en su extensión total dependerá de la zona del implante y de cada paciente. En el caso del tamaño del depósito o recámaras se definieron y evaluaron algunas dimensiones y tamaños generales. En el prototipo de implante de un depósito (a utilizar en ejemplo de aplicación 3) las medidas de 52, 2 mm de ancho y 52, 86 mm de alto para el depósito (figura 7A) . En el prototipo de implante de dos depósitos o recámaras evaluado presentaba una recámara izquierda de 24, 43 mm de ancho por 46, 7 mm de alto y un recámara o depósito izquierdo con un ancho de 24, 47 mm y 46, 84 mm de alto (figura 7B) . Las tuberías de todo el sistema tanto de entrada como de salida deben de ser de al menos 10 mm, con un máximo de 15 mm, este mínimo se debe a las pruebas desarrolladas para que el líquido pueda entrar de manera adecuada a la recámara de depósito. Ejemplo 2: Ensayo de liberación de líquido desde un implante de la presente invención Con el objetivo de estimar el tiempo de vaciado de la recámara o depósito se ha realizado un implante por impresión 3D, el que se compone de un sello de filamento de entrada de 2, 0 mm para bloquear la salid del líquido, una tubería de entrada con un sistema de embudo con un diámetro de entrada de 1, 5 mm para el ingreso de una jeringa y un diámetro posterior o de salida de 3, 0 mm. El implante probado tiene una cámara o depósito de capacidad de 5 mL con paredes de 2, 5 mm de espesor o grosor. La tubería de salida presenta un orificio de salida con un sistema embudo o de liberación controlada con una tubería de diámetro de 2, 0 mm con una transición a un diámetro de salida de 1, 3 mm (figura 6 b) . En la experiencia, el implante se llenó con 5, 1 mL de líquido en su interior, donde dicho líquido de prueba fue una solución de 4 partes de agua con una parte de azul de metileno. El protocolo consistió en inyectar los 5, 1 mL de la solución en la entrada del implante ya definido y sumergir el implante en un recipiente con agua, conservando la disposición geométrica de como seria instalado el implante. Luego, el implante se dejó en ese recipiente por un tiempo indeterminado, corroborando cada 1 hora si el color del agua ha cambiado con el colorante. Para las condiciones de prueba, se realizó la experiencia en un estado estático, es decir, sin movimiento del implante, o de forma dinámica simulando el movimiento de la cabeza del paciente. Esto último permite revisar si esto influye en la velocidad de la salida del líquido desde el interior del implante. Se observó que en la primera hora el líquido colectado desde el recipiente era casi transparente con una tonalidad o coloración celeste muy leve. A las 2 y 3 horas se observa el aumento de la tonalidad celeste como una tonalidad celeste suave. Luego de 4 horas se observa una tonalidad celeste fuerte indicando la salida del líquido introducido en el implante. Finalmente, con las pruebas realizadas hasta este momento el tiempo de vaciado sin movimiento es de aproximadamente 6 horas (ver figura 8 y tabla 2) . Cabe destacar que transcurrido este tiempo o más llegando a un límite de una semana, si el sistema se mueve el implante aún sigue liberando solución. Tabla 2.- Resultados observados experiencia liberación prolongada de líquido desde implante. Ejemplo 3: Prueba de verificación del tiempo de vaciado en implante de un depósito Se realizaron múltiples pruebas para verificar el tiempo de vaciado de la cápsula de 5 mL que posee el implante de prueba de una recámara. Se realizan además ensayos en un modo estático y en movimiento. Para los análisis se estandarizó y preparó un implante con las siguientes características técnica: • Diámetro de tapón de 2, 2 mm • Diámetro de inserción 1 mm (diámetro para insertar aguja) • Sistema embudo para disminuir la salida de líquido por la entrada en el implante • Capsula de 5mL • Paredes de al menos 2, 5 mm de grosor en todas las zonas de la cápsula o depósito • Salida de 1, 4 mm • Salida en forma de embudo para aumentar el tiempo de vaciado de cápsula o depósito Ensayo estático El experimento en su primera etapa consistió en llenar la cápsula o depósito del implante con una solución de azul de metileno diluido con agua en relación 5:1, este líquido es insertado dentro del implante verificando que la totalidad de la capacidad de la recámara o cápsula sea llenada. Luego en un recipiente plástico se procede a llenar con agua, se inserta el implante con el líquido en su interior y se ancla a un par de enganches metálicos dispuestos en el recipiente plástico para mantener una posición vertical, asemejando como este dispositivo estará posicionado en la realidad. Se evaluaron los siguientes tiempos: 24h posterior al inicio del experimento (primera muestra) , 72h posterior al inicio del experimento (segunda muestra) , 96h posterior al inicio del experimento (tercera muestra) , 120h posterior al inicio del experimento (cuarta muestra) , 144h posterior al inicio del experimento (quinta muestra) , 168 horas posterior al inicio del experimento (sexta muestra) . Los resultados de la observación de las muestras obtenidas en cada punto de tiempo permiten indicar que existe a simple vista un evidente aumento de la concentración del líquido liberado de forma prolongada al ambiente, en este caso al recipiente (figura 9A y 9B) . En la tabla 3 se describen los resultados de observación de las muestras obtenidas en cada punto de tiempo. En el tiempo de 24 horas se observó muy poca coloración del medio o salida del líquido desde el implante. En horas intermedias de 72, 96 y 120 horas se observa mayor liberación del líquido con una tonalidad celeste suave observada en el ambiente (recipiente) para que a las 144 horas la tonalidad cambie a una tonalidad celeste fuerte. Cuando se evaluó el tiempo de 168 horas se observa una mucho mayor liberación del contenido desde el reservorio o cápsula dentro del implante al medio (recipiente) . Cabe destacar que al día 11 aún sigue existiendo un goteo débil en la salida del implante, que sigue cambiando la concentración de azul de metileno en el agua del recipiente plástico. Tabla 3.- Resultados prueba de verificación del tiempo de vaciado en implante de un depósito o cápsula. Ensayo dinámico (implante en movimiento) Luego de realizar la prueba en un medio estático, se implementó una nueva prueba cuyo objetivo es recrear de manera más precisa cómo este dispositivo médico de tipo implante se comportará cuando sea usado por un paciente. En ese sentido, se recrearon los movimientos propios que ocurren en una persona (girar la cabeza, levantarse, caminar) . Para esto se utilizó una maquina 3D que tiene la capacidad de desplazar su plataforma en sentido horizontal. Sobre dicha máquina se posicionó el recipiente que contiene el agua y el implante relleno con la solución de azul de metileno anteriormente descrita en el ejemplo 2. El tipo de implante utilizado en la prueba también corresponde a un implante con las características descritas en el ejemplo de aplicación 2. Características del experimento: • Distancia de desplazamiento: 23 centímetros • Velocidad de desplazamiento: 3, 3 (cm/s) o 0, 1188 (km/h) • Ciclo corresponde al movimiento desde la posición inicial pasando por la posición final (23 centímetros) volviendo a la posición inicial, distancia recorrida 46 centímetros. • Cantidad de ciclos por minuto: 3. • Periodo de funcionamiento: 48 h. Luego de transcurridas 48h se detuvo la prueba. Transcurrido este tiempo, se observó que la concentración de azul de metileno en el medio a las 48 horas es mayor que lo observado para el tiempo 72 h en la experiencia sin movimiento (ejemplo 2) . Ejemplo 4: Implante craneofacial para evitar infecciones post-implante Luego de las iteraciones planteadas en el ejemplo de aplicación 1, se determinaron las mejores condiciones, tamaños y dimensiones del implante para poder incluir y liberar de forma gradual y correcta un líquido de interés (antibiótico y/o analgésico) . Se provee un implante craneofacial de material Poliéter éter cetona (PEEK) para evitar y disminuir las infecciones postoperación en el sitio del implante, porque dicho implante comprende: Uno o más depósitos o recámaras interiores (1) ovaladas con paredes de 2, 5 a 4 mm de grosor, que almacenan un líquido o sustancia activa en formato líquido para ser liberado de forma gradual y prolongada al sitio del implante, tuberías (2) que interconectan los depósitos o recámaras interiores con los orificios de entrada y salida, definiéndose: a) una tubería de entrada que conecta el orifico de entrada (3) con el depósito (1) de tal forma que incluye un sistema de embudo que incluye una entrada o carga del líquido que tiene un diámetro de 1, 5-2, 0 mm y que luego se ensancha en una tubería de diámetro 2, 0 a 3, 0 mm para continuar con la tubería hasta el depósito. b) una tubería de salida que conecta el depósito o recámara (1) con el orificio de salida (4) que incluye un sistema de embudo de transición que presenta un diámetro de 0, 7 a 2 mm en la parte superior y un diámetro de 0, 7 a 1, 5 mm en su parte inferior, dependiendo del diámetro del orificio de salida (4) , Uno o más orificios de entrada (3) en la parte superior del implante dependiendo del número de depósitos, donde dicho orificio es la entrada o carga del líquido para llegar a los depósitos (1) , donde el orificio de entrada tiene un diámetro de 1, 5-2, 0 mm y está bloqueado por un tapón de filamento PEEK o un tornillo de titanio, Uno o más orificios de salida (4) en la parte inferior del implante dependiendo del número de depósitos como orificio de salida gradual y prolongada del líquido o sustancia activa hacia el sitio del implante, donde el orificio de salida tiene un diámetro de 0, 7 a 1, 5 mm y tiene un tapón o sistema que sella el implante para no permitir que el líquido se escape cuando no se ha dispuesto en el sitio del implante. El implante integrado en el cráneo del paciente se acopla liberando de forma prolongada y continua el líquido contenido en los depósitos o recámaras, gracias al efecto de la gravedad y diseño del implante. Las salidas de las recámaras liberan hacia el interior del cerebro el líquido (figura 10A y 10B) . Ejemplo 5: Estudio analítico de la liberación de vancomicina esde el implante craneofacial En este estudio se revisó y evaluó la liberación de una solución de vancomicina desde el reservorio de un implante craneofacial parte del alcance de la invención. Este ensayo fue realizado en una incubadora que se encontraba a 37 grados centígrados y que realizaba un movimiento de 20 rpm, esto para imitar el movimiento que tiene un paciente luego de este tipo de cirugía. Para detectar la liberación y concentración de la solución de vancomicina liberada desde el implante, se utilizó Cromatografía Líquida de Alta Eficiencia (HPLC) acoplado a detector PDA. Como estándar se utilizó vancomicina clorhidrato con una concentración nominal de 0, 01 mg/mL preparado en suero fisiológico. Se generó una curva de calibración y la determinación de los tiempos de retención y valores de área de cada pico que aparezca en el cromatograma para determinar la concentración de vancomicina v/s el estándar. Para los muestreos como tal de la solución liberada desde el implante, se tomó una muestra de 0, 5 mL y se diluyeron en 0, 1 mL de suero fisiológico para luego llevar a un matraz de 100 mL con buffer fosfato dibásico de potasio 0, 2M a pH 7, 4. Esto se inyecta en el equipo para su análisis. Con los tiempos de retención y la determinación del área se determinó la concentración de vancomicina en la alícuota de análisis y se calculó la cantidad de vancomicina total presente en el vaso de forma acumulada (g en vaso) . Se realizaron mediciones a las 1, 2, 4, 8 horas, los resultados de estas mediciones se exponen en la tabla 4. Tabla 4.- Área detectada y cantidad de vancomicina de acuerdo tiempo de muestreo. Con este muestreo y calibración inicial se realizó un análisis de la cantidad de vancomicina liberada por tramos de tiempo. En la tabla 5 se presenta la cantidad de ancomicina liberada (g) desde la prótesis de acuerdo con el tramo de tiempo evaluado. En la figura 11, se presenta la gráfica representativa de esta cinética de liberación. De acuerdo con los resultados, en el tramo de la primera hora se observa la mayor liberación de vancomicina desde el implante. Desde la hora 2 en adelante, se observa que la liberación aumenta sostenida y paulatinamente con el avance del tiempo. Esta cinética de liberación permite en un inicio la exposición de una mayor cantidad del principio activo en la zona con el fin de alcanzar rápidamente concentraciones terapéuticas y luego la regulación de la salida del principio activo de forma paulatina en el tiempo. Si nos referimos a la tasa de liberación, en la primera hora se observa una alta tasa de liberación de vancomicina con un promedio de 218 g/mL, la que luego disminuye en el tramo de tiempo entre la primera y segunda hora, para luego aumentar y mantener la liberación sostenida. En la figura 12 se presenta la gráfica representativa de la tasa de liberación de vancomicina desde el implante en los tiempos de muestreo expuestos en la tabla 5. Tabla 5.- Cantidad de vancomicina liberada y tasa de liberación de ésta. A partir de los muestreos de los tiempos 2, 4, 8 y 12 horas se determinó la función o ecuación de la recta que permite predecir cuánto tiempo el implante seguirá liberando el principio activo: f (x) = 911, 6x+11.465, 1. Con esta información se extrapoló y determinó teóricamente la cantidad de vancomicina liberada en horas posteriores. En la tabla 6 se exponen las cantidades de vancomicina liberada acumulada y definida para los tiempos de 24, 36 y 43 horas. Esta simulación o considera que luego de las 12 horas, la velocidad podría disminuir, y por lo tanto, aumentar el tiempo de entrega. Tabla 6.- Cantidad de vancomicina liberada acumulada y determinada hasta las 43 horas. Ejemplo 7: Estabilidad principio activo en solución dentro del implante. Para este análisis se consideró nuevamente trabajar con el principio activo vancomicina, sin limitarse a otros principios activos. Con este ensayo se busca demostrar que la vancomicina es establece dentro del implante y no se degrada mientras es contenido, por lo tanto, su liberación es funcional. Para esto se analizó una muestra a tiempo cero (To) correspondiente a una muestra analizada inmediatamente luego de su preparación y una muestra que se dejó durante 24 horas en el implante. De esta forma, se compararon los resultados del tiempo 24 horas con el tiempo cero, como áreas de muestra y porcentaje de degradación a las 24 horas. La preparación de la solución muestra al tiempo cero (T0) consistió en tomar un frasco ampolla de vancomicina de 500 mg y adicionar 5 mL de suero fisiológico. Agitar la solución completa del liofilizado, tomar 0, 5mL de dicha ampolla, llevar a 5 mL de volumen final con suero fisiológico. Se toman estos 5mL y se inyectan en el depósito del implante. En el caso de la muestra a las 24 horas, se toma 0, 5 mL de una solución desde una ampolla de vancomicina de 500mg reconstituida en 5 mL de suero fisiológico. Los 0, 5 mL son llevados a 5mL de volumen final con buffer fosfato dibásico de potasio 0, 2 M pH 7, 4. Luego, con una jeringa se carga dicho volumen en el depósito o cápsula del implante, se sella el implante y se incorpora en un vaso precipitado vacío. La concentración nominal de la solución en la cámara es de 10 mg/mL. Luego de las 24 horas, se abrió la cápsula o depósito para extraer 0, 5 mL de la solución contenida en su interior. De los 0, 5 mL extraídos desde el depósito se toma una alícuota de 0, 1 mL que se llevan a un matraz de 100 mL para su análisis por HPLC con la misma curva de calibración y estándar descrito para el ejemplo anterior. En la tabla 7 se exponen los resultados de este ensayo. Se observa que a las 24 horas la degradación de la vancomicina no es significativa. Estos resultados demuestran que el principio activo incluido en el o los depósitos del implante de la presente invención se mantiene estable para su liberación. Tabla 7.- Resultados de ensayos de estabilidad de vancomicina en implante de la presente invención. Bibliografía (Kwarcinski, Jeremy & Boughton, Philip & Ruys, Andrew & Doolan, Alessandra & van Gelder, James. (2017) . Cranioplasty and Craniofacial Reconstruction: A Review of Implant Material, Manufacturing Method and Infection Risk. Applied Sciences. 7. 276. 10.3390/app7030276) . Chen Y, Zhang L, Qin T, Wang Z, Li Y, Gu B. (2019) . Evaluation of neurosurgical implant infection rates and associated pathogens: evidence from 1118 postoperative infections. Neurosurg Focus. 47 (2) :E6. doi: 10.3171/2019.5.FOCUS18582. Chengzhe Gao, Zongliang Wang, Zixue Jiao, Zhenxu Wu, Min Guo, Yu Wang, Jianguo Liu, Peibiao Zhang. (2021) . Enhancing antibacterial capability and osseointegration of olyetheretherketone (PEEK) implants by dual-functional surface modification, Materials & Design, Volume 205, 109733. https://doi.org/10.1016/j.matdes.2021.109733 Delaney, LJ, MacDonald, D., Leung, J., Fitzgerald, K., Sevit, AM, Eisenbrey, JR, Patel, N., Forsberg, F., Kepler, CK, Fang, T., Kurtz, SM, & Hickok, Nueva Jersey (2019) . Liberación de antibióticos activada por ultrasonido de los clips de PEEK para prevenir la infección por fusión espinal: evaluaciones iniciales. Acta biomaterialia, 93, 12-24. https://doi.org/10.1016/j.actbio.2019.02.041 Gu Xinming, Sun Xiaolin, Sun Yue, Wang Jia, Liu Yiping, Yu Kaixuan, Wang Yao, Zhou Yanmin. (2021) . Bioinspired Modifications of PEEK Implants for Bone Tissue Engineering. Frontiers in Bioengineering and Biotechnology, Vol. 8, doi:10.3389/fbioe.2020.631616. Hernandez Cantu, Enoc Isaí; Esparza Dávila, Sandra Paloma; Reyes Silva, Alan Karim Sayeg. (2020) . Eficacia de un modelo de prevención de infección de sitio quirúrgico en un hospital de segundo nivel de atención. Index Enferm, Granada, v. 29, n. 1-2, p. 9 12. Lazar, M. A., Vodnar, D., Prodan, D., Rotaru, H., Roman, C. R., Sorcoi, L. A., Baciut, G.; Campian, R. S. (2016) . Antibacterial coating on biocomposites for cranio-facial reconstruction.Clujul medical (1957) , 89 (3) , 430-434. https://doi.org/10.15386/cjmed-599 MINSAL. Ministerio de Salud de Chile. Departamento de Calidad y Seguridad en la atención, Programa de control de IAAS. (2017) . Informe de vigilancia de infecciones asociadas a la atención de salud. https://www.minsal.cl/wpcontent/uploads/2015/09/informe-vigilancia-2017.pdf Xue, Z., Wang, Z., Sun, A., Huang, J., Wu, W., Chen, M., et al. (2020) . Rapid construction of polyetheretherketone (PEEK) biological implants incorporated with brushite (CaHPO4.2H2O) and antibiotics for anti-infection and enhanced osseointegration. Mater. Sci. Eng. C Mater. Biol. Appl. 111:110782. doi: 10.1016/j.msec.2020.110782 Yan, J., Zhou, W., Jia, Z., Xiong, P., Li, Y., Wang, P., et al. (2018) . Endowing polyetheretherketone with synergistic bactericidal effects and improved osteogenic ability. Acta Biomater. 79, 216-229. doi: 10.1016/j.actbio.2018.08.037
Publicaciones:
ES2957984 (30/01/2024) - A1 Solicitud de patente con informe sobre el estado de la técnica
Eventos:
En fecha 20/01/2023 se realizó Registro Instancia de Solicitud
En fecha 20/01/2023 se realizó Admisión a Trámite
En fecha 20/01/2023 se realizó 1001P_Comunicación Admisión a Trámite
En fecha 23/01/2023 se realizó Superado examen de oficio
En fecha 21/11/2023 se realizó Realizado IET
En fecha 24/11/2023 se realizó 1109P_Comunicación Traslado del IET
En fecha 30/01/2024 se realizó Publicación Solicitud
En fecha 30/01/2024 se realizó Publicación Folleto Solicitud con IET (A1)
En fecha 18/04/2024 se realizó PETEX_Petición de examen sustantivo
En fecha 18/04/2024 se realizó PETEX_Petición de examen sustantivo
En fecha 18/04/2024 se realizó 5215P_Observaciones del solicitante al IET, Opinión Escrita y/o alegaciones a observaciones de terceros
Pagos:
20/01/2023 - Pago Tasas IET
Fuente de la información
Parte de la información aquí publicada es pública puesto que ha sido obtenida de la Oficina de Propiedad Industrial de los diferentes países el 08/05/2024 y por lo tanto puede ser que la información no esté actualizada.Parte de la información aquí mostrada ha sido calculada por nuestro sistema informático y puede no ser veraz.
Privacidad
Si considera que al información aquí publicada afecta a su privacidad y desea que eliminemos la información aquí publicada envíe un email a info@patentes-y-marcas.com o rellene el formulario que encontrará aquí.Información sobre el registro de patente nacional por IMPLANTE CRANEOFACIAL DE POLIÉTER ÉTER CETONA (PEEK) CON DEPÓSITOS PARA ALMACENAR Y LIBERAR SUSTANCIAS ACTIVAS con el número P202330037
El registro de patente nacional por IMPLANTE CRANEOFACIAL DE POLIÉTER ÉTER CETONA (PEEK) CON DEPÓSITOS PARA ALMACENAR Y LIBERAR SUSTANCIAS ACTIVAS con el número P202330037 fue solicitada el 20/01/2023. Se trata de un registro en España por lo que este registro no ofrece protección en el resto de países. El registro IMPLANTE CRANEOFACIAL DE POLIÉTER ÉTER CETONA (PEEK) CON DEPÓSITOS PARA ALMACENAR Y LIBERAR SUSTANCIAS ACTIVAS con el número P202330037 fue solicitada por ARCOSYSTEM SPA mediante los servicios del agente Nuria Capitan García. El registro [modality] por IMPLANTE CRANEOFACIAL DE POLIÉTER ÉTER CETONA (PEEK) CON DEPÓSITOS PARA ALMACENAR Y LIBERAR SUSTANCIAS ACTIVAS con el número P202330037 está clasificado como A61F 2/28 según la clasificación internacional de patentes.
Otras invenciones solicitadas en la clasificación internacional de patentes A61F 2/28.
Es posible conocer invenciones similares al campo de la técnica se refiere. El registro de patente nacional por IMPLANTE CRANEOFACIAL DE POLIÉTER ÉTER CETONA (PEEK) CON DEPÓSITOS PARA ALMACENAR Y LIBERAR SUSTANCIAS ACTIVAS con el número P202330037 está clasificado con la clasificación A61F 2/28 por lo que si se desea conocer más registros con la clasificación A61F 2/28 clicar aquí.Otras invenciones solicitadas a través del representante NURIA CAPITAN GARCÍA
Es posible conocer todas las invenciones solicitadas a través del agente NURIA CAPITAN GARCÍA entre las que se encuentra el registro patente nacional por IMPLANTE CRANEOFACIAL DE POLIÉTER ÉTER CETONA (PEEK) CON DEPÓSITOS PARA ALMACENAR Y LIBERAR SUSTANCIAS ACTIVAS con el número P202330037. Si se desean conocer más invenciones solicitadas a través del agente NURIA CAPITAN GARCÍA clicar aquí.Patentes en España
Es posible conocer todas las invenciones publicadas en España entre las que se encuentra el registro patente nacional por IMPLANTE CRANEOFACIAL DE POLIÉTER ÉTER CETONA (PEEK) CON DEPÓSITOS PARA ALMACENAR Y LIBERAR SUSTANCIAS ACTIVAS. Nuestro portal www.patentes-y-marcas.com ofrece acceso a las publicaciones de patentes en España. Conocer las patentes registradas en un país es importante para saber las posibilidades de fabricar, vender o explotar una invención en España.Patentes registradas en la clase A
Es posible conocer todas las patentes registradas en la clase A (NECESIDADES CORRIENTES DE LA VIDA) entre las que se encuentra la patente IMPLANTE CRANEOFACIAL DE POLIÉTER ÉTER CETONA (PEEK) CON DEPÓSITOS PARA ALMACENAR Y LIBERAR SUSTANCIAS ACTIVAS con el número P202330037. Conocer las patentes registradas en una clase es importante para saber las posibilidades de registrar una patente en esa misma clase.Patentes registradas en la clase A61
Es posible conocer todas las patentes registradas en la clase A61 (CIENCIAS MEDICAS O VETERINARIAS; HIGIENE) entre las que se encuentra la patente IMPLANTE CRANEOFACIAL DE POLIÉTER ÉTER CETONA (PEEK) CON DEPÓSITOS PARA ALMACENAR Y LIBERAR SUSTANCIAS ACTIVAS con el número P202330037. Conocer las patentes registradas en una clase es importante para saber las posibilidades de registrar una patente en esa misma clase.Patentes registradas en la clase A61F
Es posible conocer todas las patentes registradas en la clase A61F (FILTROS IMPLANTABLES EN LOS VASOS SANGUINEOS; PROTESIS; DISPOSITIVOS QUE MANTIENEN LA LUZ O QUE EVIT) entre las que se encuentra la patente IMPLANTE CRANEOFACIAL DE POLIÉTER ÉTER CETONA (PEEK) CON DEPÓSITOS PARA ALMACENAR Y LIBERAR SUSTANCIAS ACTIVAS con el número P202330037. Conocer las patentes registradas en una clase es importante para saber las posibilidades de registrar una patente en esa misma clase.
¿Tienes alguna duda?
Escribe tu consulta y te responderemos rápida y gratuitamente.

Otras patentes similares
 P202300069
P202300069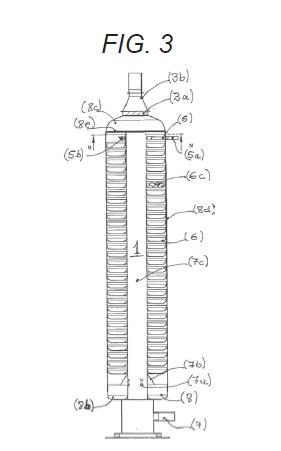 P202330068
P202330068 P202330089
P202330089Profesionales Recomendados
Barcelona
933182440
España
933182440
España

Barcelona
+34 93 362 16 97
España
+34 93 362 16 97
España

Barcelona
932 593 600
España
932 593 600
España







